Czerniak (nowotwór)
| melanoma malignum | |
 Czerniak szerzący się powierzchownie | |
| ICD-10 | C43 |
|---|---|
| C43.0 | Czerniak wargi |
| C43.1 | Czerniak powieki, łącznie z kątem oka |
| C43.2 | Czerniak ucha i przewodu słuchowego zewnętrznego |
| C43.3 | Czerniak innych i nieokreślonych części twarzy |
| C43.4 | Czerniak owłosionej skóry głowy i skóry szyi |
| C43.5 | Czerniak tułowia |
| C43.6 | Czerniak kończyny górnej, łącznie z barkiem |
| C43.7 | Czerniak kończyny dolnej, łącznie z biodrem |
| C43.8 | Czerniak skóry przekraczający granice jednego umiejscowienia |
| C43.9 | Czerniak skóry, umiejscowienie nieokreślone |
| ICDO | 8720/3 |
Czerniak, zwany też czerniakiem złośliwym[a] (łac. melanoma malignum) – nowotwór złośliwy skóry, błon śluzowych lub błony naczyniowej oka wywodzący się z melanocytów. Stanowi od 5% do 7% nowotworów złośliwych skóry człowieka. Corocznie jest rozpoznawane około 130 000 przypadków tej choroby, większość u osób rasy białej. Zapadalność na świecie wykazuje znaczną zależność geograficzną. Czerniak przed 40. rokiem życia jest rzadki, szczyt zachorowań przypada na siódmą i ósmą dekadę życia.
Najczęściej pojawia się na skórze niezmienionej, choć może powstać w obrębie znamion barwnikowych. Podejrzenie czerniaka budzi pojawienie się nowej zmiany przypominającej atypowe znamię lub zmiana wcześniej istniejącego znamienia barwnikowego. Do głównych objawów czerniaka należą: asymetryczne zabarwienie, kształt i powierzchnia zmiany, uniesienie się zmiany ponad otaczającą skórę, nieregularne ograniczenie zmiany, a także duży jej wymiar. Również świąd, ból, krwawienie i owrzodzenie w obrębie znamienia lub nowej zmiany skórnej budzą podejrzenie złośliwego charakteru zmiany.
Głównymi czynnikami ryzyka są ekspozycja na promieniowanie ultrafioletowe pochodzące z promieniowania słonecznego lub sztucznych źródeł i indywidualna podatność na nowotwór, przede wszystkim przez zwiększone ryzyko genetyczne. Intensywna, przerywana ekspozycja na promieniowanie ultrafioletowe prowadząca do oparzenia słonecznego, szczególnie w okresie dzieciństwa, wiąże się z większym ryzykiem zachorowania niż wieloletnia regularna ekspozycja. Nowotwór częściej występuje u kobiet niż u mężczyzn. Rozpoznanie kliniczne jest stawiane na podstawie badania dermatoskopowego, a pozostałe badania pełnią rolę jedynie pomocniczą.
Podejrzana zmiana jest wycinana z niewielkim marginesem zdrowych tkanek, a próbka jest badana histopatologicznie. Badanie to umożliwia ostateczne rozpoznanie choroby oraz ocenę wielu czynników o charakterze rokowniczym. Podstawową metodą leczenia czerniaka jest radykalne wycięcie guza z odpowiednio szerokim marginesem zdrowych tkanek. Często jest konieczna biopsja węzła wartowniczego lub rozszerzenie zabiegu o wycięcie lokalnych węzłów chłonnych. W przypadku choroby nieoperacyjnej lub obecności przerzutów odległych stosuje się leczenie ogólnoustrojowe oparte na nowoczesnych lekach celowanych, dobieranych indywidualnie dla każdego chorego w oparciu o bieżące mutacje i obraz kliniczny choroby. Wykorzystywanych jest kilka grup leków celowanych: inhibitory BRAF (wemurafenib, dabrafenib), inhibitory MEK (trametynib), przeciwciała anty-CTLA-4 (ipilimumab), przeciwciała anty-PD-1 (pembrolizumab, niwolumab) i inhibitory c-KIT (imatynib). W leczeniu stosuje się również klasyczną immunoterapię opartą o interferon α i IL-2. Klasyczna chemioterapia jest stosowana u chorych nieodpowiadających na leki celowane i immunoterapię. Zapobieganie polega na unikaniu nadmiernego wystawienia się na działanie promieniowania słonecznego.
Objawy
W postaci wczesnej czerniak jest płaską, niesymetryczną zmianą o nieregularnych, poszarpanych granicach ze zdrową skórą i nieregularnym zabarwieniu. W późniejszym stadium nowotwór ulega uniesieniu ponad otaczającą skórę. Mogą pojawić się owrzodzenie i krwawienie ze zmiany. Pierwszymi objawami choroby nowotworowej mogą być powiększenie węzłów chłonnych lub objawy związane z przerzutami[1].
Czerniak w większości przypadków powstaje na skórze niezmienionej (de novo) bez poprzedzających go znamion barwnikowych (tzw. „pieprzyki”)[2], może również pojawić się w obrębie łagodnych znamion barwnikowych, szczególnie znamion dysplastycznych. Podejrzenie czerniaka mogą nasuwać nowe zmiany skórne przypominające nietypowe znamiona oraz zmiany wcześniej występujących znamion barwnikowych, szczególnie zgrubienie tych zmian, zmiana ich kształtu i granic, zmiana zabarwienia, a także pojawienie się swędzenia, bólu, krwawienia lub owrzodzenia[3].
Tradycyjnie objawy kliniczne grupuje się w system ABCD(E)[4]:
- A (asymmetry) – asymetria – czerniak w odróżnieniu od zmian barwnikowych wykazuje nieregularny wygląd,
- B (borders) – granice – brzegi zmiany są nierówne i postrzępione,
- C (color) – kolor – kolor czerniaka jest różnorodny od jasnobrązowego do czarnego, nierównomiernie rozłożony, często z punktowym skupieniem barwnika,
- D (diameter) – średnica zmiany > 5 mm,
- E (elevate) – uwypuklenie – uwypuklenie się zmiany ponad otaczający naskórek.
Większość znamion barwnikowych jest łagodna i niegroźna. Roczne ryzyko przemiany złośliwej jest niskie i wynosi dla pojedynczej zmiany 1:200 000[2][5]. Łagodne znamię barwnikowe jest jednolite pod względem zabarwienia, koloru brązowego lub czarnego, jest ostro odgraniczone od normalnej skóry, owalnego lub okrągłego kształtu, o wielkości nieprzekraczającej 5 mm. Oceny lekarskiej za pomocą dermatoskopu wymagają znamiona zmieniające swój wygląd oraz znamiona nietypowe i wrodzone[6].
- Zespoły paraneoplastyczne
Czerniakowi bardzo rzadko mogą towarzyszyć niecharakterystyczne zespoły paraneoplastyczne[7][8]:
- skórne – zapalenie skórno-mięśniowe, bielactwo nabyte, twardzina układowa, pęcherzyca paraneoplastyczna, melanoza, acanthosis nigricans
- oczne – retinopatia związana z czerniakiem (ang. melanoma-associated retinopathy)[9]
- hematologiczne – reakcja leukemiczna, eozynofilia, neutropenia
- metaboliczne – hiperkalcemia, zespół Cushinga, osteoartropatia przerostowa
- neurologiczne – przewlekła demielinizacyjna polineuropatia.
Czynniki ryzyka

Do czynników sprzyjających wystąpieniu czerniaka należą[10]:
- ekspozycja na promieniowanie słoneczne (naturalne promieniowanie UV)
- duża skumulowana ekspozycja na promieniowanie słoneczne
- sporadyczna, przerywana i intensywna ekspozycja na światło słoneczne, oparzenia słoneczne w dzieciństwie i wieku młodzieńczym
- sztuczna ekspozycja na promieniowanie ultrafioletowe (solaria, opalanie się, lampy do utwardzania lakieru w manicure hybrydowym[11])
- jatrogenna ekspozycja na promieniowania ultrafioletowe wraz z psoralenem – fotochemioterapia (PUVA)
- występowanie czerniaka u krewnych pierwszego i drugiego stopnia
- wcześniejsze występowanie czerniaka u tego samego chorego – ryzyko zwiększone 8–10 razy[12]
- występowanie innego nieczerniakowego nowotworu złośliwego skóry, w tym raka podstawnokomórkowego, raka kolczystokomórkowego[13]
- zespół znamion dysplastycznych
- duża liczba znamion barwnikowych (melanocytowych) oraz dużych wrodzonych znamion barwnikowych
- jasna karnacja skóry, jasny kolor włosów i oczu, obecność piegów, łatwe uleganie oparzeniom słonecznym[13][12][14]
- wiek – ryzyko zachorowania wzrasta wraz z wiekiem
- skóra pergaminowa (xeroderma pigmentosum) – stukrotnie zwiększone ryzyko[13]
- wysoki status socjoekonomiczny[15]
- immunosupresja i przeszczepienie narządu[12]
- prawdopodobnie promieniowanie jonizujące[16].
Ekspozycja na promieniowanie ultrafioletowe
Ekspozycja na działanie promieniowania ultrafioletowego jest uważana za główny środowiskowy czynnik ryzyka wystąpienia czerniaka[17][10]. Największe ryzyko niesie promieniowanie o zakresie długości fali 280–320 nm zaliczane do UV-B[10][18][19]. Ryzyko jest w różny sposób związane z rodzajem ekspozycji na promieniowanie ultrafioletowe, a stopień ryzyka zależy od dawki, długości ekspozycji (narażenie przerywane i przewlekłe) i wieku w czasie ekspozycji[17].
Największe zagrożenie czerniakiem niosą krótkie, przerywane epizody narażenia na naturalne lub sztuczne promieniowanie ultrafioletowe, szczególnie w dzieciństwie, wielokrotne oparzenia słoneczne oraz wysokie regularne narażenie na słońce przez całe życie[17][10][20][21].
Przerywana intensywna ekspozycja na światło słoneczne i promieniowanie ultrafioletowe skutkuje większym ryzykiem zachorowania niż regularne narażenie w życiu dorosłym[22]. Wykazano, że imigranci z obszarów o dużej ekspozycji na promieniowanie ultrafioletowe i o jasnej karnacji do obszarów o niskim narażeniu wciąż mają podwyższone życiowe ryzyko czerniaka[23]. Uważa się, że unikanie nadmiernego narażenia na słońce w dzieciństwie ma większy wpływ na ryzyko zachorowania niż unikanie go w dorosłości[21]. Ekspozycja w życiu dorosłym ma mniejsze znaczenie, jednak również w pewnym stopniu wpływa na zwiększone ryzyko czerniaka. Badania wskazują, że imigranci z obszarów o niskim ryzyku zachorowania na czerniaka osiedlający się w regionach o wysokim ryzyku wykazują wyższą częstość zachorowania niż mieszkańcy obszarów, z których pochodzą[24]. Sugeruje to nakładanie się ryzyka związanego z ekspozycją w dzieciństwie z ekspozycją w wieku dorosłym i wzrost ryzyka zachorowania[23]. W regionach o niskiej zapadalności na czerniaka większe ryzyko zachorowania jest związane z przerywaną ekspozycją i oparzeniami słonecznymi niż regularnym wystawieniem na działanie promieniowania ultrafioletowego. Przewlekły wzór ekspozycji jest bardziej związany z regionami o wysokim ciągłym narażeniu na promieniowanie ultrafioletowe[25][23].
Stosowanie sztucznych źródeł promieniowania ultrafioletowego, takich jak solaria i lampy opalające, zwiększa względne ryzyko czerniaka. Młody wiek początku korzystania z solarium jest silnie związany ze zwiększonym ryzykiem zachorowania[26][27].
Znamiona barwnikowe i znamiona dysplastyczne
.jpg)
.jpg)
.jpg)
Do czynników ryzyka zachorowania na czerniaka należy duża ilość znamion barwnikowych (melanocytowych), znamiona dysplastyczne oraz duże (> 20 cm średnicy) znamiona wrodzone (tzw. znamiona kąpielowe)[28][29].
- Łagodne znamiona barwnikowe
Większość znamion barwnikowych jest łagodna i niegroźna. Roczne ryzyko przemiany złośliwej jest niewielkie i dla pojedynczej zmiany wynosi 1:200 000[2][5]. Łagodne znamię barwnikowe jest jednolite pod względem zabarwienia, jest koloru brązowego lub czarnego, ostro odgraniczone od normalnej skóry, owalnego lub okrągłego kształtu, o wielkości nieprzekraczającej 5 mm. Oceny lekarskiej za pomocą dermatoskopu wymagają znamiona zmieniające swój wygląd bądź znamiona nietypowe i wrodzone[6].
Wykazano, że wysoka liczba znamion barwinkowych jest czynnikiem ryzyka czerniaka. Wysoką liczbę znamion barwnikowych często definiuje się przez obliczenie liczby znamion na plecach, których ilość przekracza 40. 40% czerniaków rozwija się w obrębie lub sąsiedztwie znamion barwnikowych[14][30][31]. Na ryzyko związane z dużą liczbą łagodnych znamion barwnikowych wpływa ekspozycja na promieniowanie ultrafioletowe (w tym światło słoneczne) i osobnicza predyspozycja do rozwoju tego nowotworu[32]. Podwyższone ryzyko nowotworu prawdopodobnie wynika z losowych zmian genetycznych w obrębie melanocytów tworzących znamiona, które ostatecznie doprowadzają do rozwoju nowotworu złośliwego. Ryzyko powstania czerniaka jest jednak bardzo małe w porównaniu do ogromnej liczby łagodnych znamion barwnikowych w populacji. Nie ma uzasadnienia dla wycinania łagodnych znamion barwnikowych bez klinicznych cech dysplazji[32].
- Znamiona dysplastyczne
Znamię dysplastyczne jest to wariant łagodnego znamienia barwnikowego z atypią cytologiczną i architektoniczną (histopatologiczną)[32]. Klinicznie znamię dysplastyczne cechuje się nieregularną granicą, asymetrią, nierównomiernym zabarwieniem o różnym odcieniu brązu z elementami z odcieniami barwy różowej zwykle w obrębie obręczy zmiany i wielkości przekraczającej 5 mm. Do rozpoznania konieczna jest obecność płaskich elementów w znamieniu i stwierdzenie wielkości powyżej 5 mm[33]. Znamiona dysplastyczne występują dość powszechnie: u około 2–8% populacji[12][28].
Obecność znamion dysplastycznych zwiększa ryzyko zachorowania[12], jednak zdecydowana większość takich zmian nie ulega progresji do czerniaka[34]. Ryzyko rozwoju czerniaka na bazie znamiona dysplastycznego wynosi 1:3000 na rok[35]. Ich obecność jest przede wszystkim ważnym markerem podwyższonego ryzyka zachorowania[12][28]. Na faktyczne ryzyko wpływają inne czynniki ryzyka, głównie ekspozycja na promieniowanie ultrafioletowe[36]. Sama obecność znamion dysplastycznych nie jest uważana za prekursora wysokiego ryzyka czerniaka, a raczej za marker wskazujący na indywidualnie podwyższone ryzyko czerniaka[35][33].

- Zespół znamion dysplastycznych
Zespół znamion dysplastycznych (zespół znamion atypowych, rodzinny zespół znamion atypowych i czerniaka, ang. familial atypical multiple mole melanoma syndrome, FAMMMS) jest to zespół predyspozycji do występowania znacznej liczby atypowych znamion i znacznie zwiększonego ryzyka rozwoju czerniaka skóry[37].
Zespół występuje sporadycznie lub występuje rodzinnie dziedziczony autosomalnie dominująco ze zmienną ekspresją i penetracją. U połowy rodzin z tym zespołem wykryto mutację genu CDKN2A kodującego p16, który pełni rolę genu supresorowego[38].
Klinicznie zespół objawia się liczbą łagodnych znamion barwnikowych przekraczającą sto, obecnością wielu znamion dysplastycznych i znacznie zwiększonym ryzykiem wystąpienia czerniaka. Znamiona występują w nietypowych lokalizacjach, w tym w obszarach nieeksponowanych na działanie słońca. Najczęściej pojawiają się w okresie dojrzewania, ale również mogą powstawać u dzieci i dorosłych. Nie ma ogólnie przyjętych kryteriów rozpoznania FAMMMS[28].
Ryzyko zachorowania na czerniaka u chorych bez występowania czerniaka w rodzinie jest 2–28-krotnie zwiększone i 150-krotnie zwiększone u chorych, u których występował czerniak w rodzinie[33]. Szacuje się, że prawdopodobieństwo zachorowania na czerniaka do 80. roku życia wynosi 60–80%. Rak trzustki jest drugim najczęściej występującym nowotworem w tym zespole, a ryzyko zachorowania 13–22-krotnie większe niż ryzyko populacyjne. Szacuje się, że ryzyko zachorowania na raka trzustki do 75. roku życia wynosi około 20%[39]. Istnieją doniesienia o innych nowotworach współwystępujących z zespołem, ale ich korelacja z zespołem znamion dysplastycznych nie jest oczywista[39][40].
Czynniki genetyczne
Występowanie czerniaka w rodzinie znacząco zwiększa ryzyko zachorowania. Stwierdzono, że występowanie czerniaka u krewnych pierwszego stopnia 2–3-krotnie zwiększa ryzyko zachorowania[41][14]. W około 10% przypadków czerniaka jest stwierdzane jego rodzinne występowanie, które jest związane z genetyczną predyspozycją na ten nowotwór[12][27]. Największą rolę pełnią gen CDKN2A (p16) zlokalizowany na chromosomie 9p21 oraz gen CDK4 zlokalizowany na chromosomie 12q14[27].
- Geny wysokiego ryzyka
Do genów wysokiego ryzyka zalicza się CDKN2A, CDKN2B i CDK4[42].
Mutacja CDKN2A jest stwierdzana w 25% przypadków rodzinnego występowania czerniaka[41]. Gen CDKN2A koduje białko p16 biorące udział w regulacji cyklu komórkowego i pełniące rolę antyonkogenu. Białko p16 jest inhibitorem kinaz zależnych od cyklin i działa poprzez związanie z CDK4. Mutacja CDKN2A niesie 30% ryzyko zachorowania do 80 roku życia[43].
Gen CDKN2B leży w pobliżu CDKN2A i wykazuje podobny mechanizm działania[44].
| Typ (synonim) | Gen | Locus | OMIM |
| CMM1 (zespół znamion dysplastycznych), zespół B-K, FAMMM, DNS | CMM1 | 1p36 | OMIM 155600 |
| CMM2 | CDKN2A | 9p21 | OMIM 155601 |
| CMM3 | CDK4 | 12q14 | OMIM 609048 |
| CMM4 | 1p22 | OMIM 608035 | |
| Polimorfizm genu MC1R | MC1R | 16q24.4 | OMIM 155555 |
| Czerniak jagodówki | UVM1 | 3q | OMIM 155720 |
| Zespół czerniak-gwiaździak | CDKN2A | 9p21 | OMIM 155755 |
| Zespół czerniak-rak trzustki (FAMMMPC) | CDKN2A | 9p21 | OMIM 606719 |
- Geny niskiego ryzyka
Gen MC1R koduje receptor dla melanotropiny (MSH) uczestniczący w wytwarzaniu melaniny przez melanocyty. Jeden z wariantów tego genu jest związany z rudym kolorem włosów, jasnym fenotypem skóry i piegami, które są z kolei cechami związanymi ze zwiększonym ryzykiem czerniaka. Kilka badań wskazuje na zwiększone ryzyko czerniaka w niektórych wariantach tego genu, a największe ryzyko stwierdzono w wariancie Asp294His[44][45].
Innymi zidentyfikowanymi genami niskiego ryzyka są geny związane z naprawą DNA, w tym rodzina genów XP (xeroderma pigmentosum: XPC[46][47][48] i XPD[48][49]) i BRCA2[50]. Również polimorfizm genu receptora witaminy D może być związany ze zwiększonym ryzykiem zachorowania[51].
Fenotyp skóry
Fenotyp jest niezależnym czynnikiem ryzyka zachorowania. Osoby z jasnym kolorem skóry, włosów, oczu i obecnością piegów mają zwiększone ryzyko zachorowania na czerniaka[14].
Status socjoekonomiczny
Czerniak jest częstszy w grupach o wyższym statusie socjoekonomicznym. Prawdopodobnie jest to związane z większą szansą rekreacyjnego opalania się, np. podczas uprawiania sportu i w miesiącach zimowych[12][15][52].
Immunosupresja
Immunosupresja zwiększa ryzyko czerniaka. Chorzy, którzy otrzymali przeszczep narządu, mają ośmiokrotnie zwiększone ryzyko zachorowania na czerniaka[12][53].
Zapobieganie
Ochrona przed słońcem
Filtry przeciwsłoneczne chronią przed rakiem kolczystokomórkowym, jednak ich wpływ na ochronę przed rakiem podstawnokomórkowym i czerniakiem jest niejasny, mimo skutecznej ochrony przeciwsłonecznej (zapobieganiu oparzeniom słonecznym)[54]. Ponadto stosowanie filtrów przeciwsłonecznych może prowadzić do przedłużonej ekspozycji na słońce i paradoksalnie zwiększyć ryzyko zachorowania[52][55].
Niektóre badania wskazują, że ochrona za pomocą filtrów przeciwsłonecznych zmniejsza ilość powstających znamion barwnikowych[56], jednak nie ma dowodów na zapobieganie czerniakowi[57]. Z drugiej strony obecne filtry przeciwsłoneczne oferują czterokrotnie większą ochronę przeciwsłoneczną niż te dostępne dziesięć lat temu, co teoretycznie może zwiększyć skuteczność ochrony przed czerniakiem[54][58].
Mimo niepełnych dowodów zaleca się stosować filtry przeciwsłoneczne ze wskaźnikiem ochrony przeciwsłonecznej przynajmniej 15[54]. Zaleca się również unikanie ekspozycji na działanie najsilniejszego promieniowania słonecznego, należy zakrywać głowę przed słońcem i chronić oczy za pomocą okularów przeciwsłonecznych oraz unikać sztucznych źródeł promieniowania ultrafioletowego, w tym solariów i lamp opalających[10][14][54].
Edukacja i samobadanie
Istotnym elementem profilaktyki jest edukacja społeczeństwa w ramach profilaktyki pierwotnej, szczególnie osób młodych, o potencjalnych czynnikach zwiększających ryzyko zachorowania i sposobach im przeciwdziałania[10]. Kolejnym elementem zapobiegania jest profilaktyka wtórna, czyli wczesne wykrywanie zmian. Popularyzacja wiedzy o czerniaku i popularyzacja samobadania skóry może pomóc zredukować śmiertelność z jego powodu. Niemal 80% chorych z czerniakiem szerzącym się powierzchownie i 70% z czerniakiem guzkowym zauważyło zmianę wyglądu znamiona barwnikowego[54]. Jednocześnie stwierdzono duże opóźnienie pomiędzy stwierdzeniem przekształcania się zmiany a poszukiwaniem porady lekarskiej, co sprzyja późniejszemu rozpoznaniu bardziej zaawansowanej i gorzej rokującej choroby[59].
Badania przesiewowe
Badania przesiewowe koncentrują się na zbadaniu całej populacji lub części populacji w ramach wybranych kryteriów włączenia do badania, w celu wczesnego wykrycia nowotworu i redukcji śmiertelności z jego powodu, co jest podstawowym celem tych badań. Obecnie nie ma wystarczających dowodów, by przeprowadzać ogólnopopulacyjne badania przesiewowe w kierunku nowotworów złośliwych skóry. Nie wykazano, by badania przesiewowe zmniejszały śmiertelność z powodu nowotworów skóry, w tym czerniaka[10][60].
Populacyjne badania przesiewowe były rozważane w Australii, w kraju o jednej z najwyższych zapadalności na czerniaka na świecie. Pilotażowe badanie przeprowadzone na części obszaru kraju cieszyło się bardzo dużą popularnością, jednak zrezygnowano z niego ze względu na jego wysoki koszt[15][61][62].
Szczepienia
Niektóre szczepienia przeciwko chorobom zakaźnym w dzieciństwie wykazują działanie ochronne przed czerniakiem. Szczepionka BCG przeciw gruźlicy oraz szczepionka przeciw ospie prawdziwej znacząco redukują ryzyko powstania czerniaka[63][64][65]. Wzrost ryzyka zachorowania na czerniaka w niektórych krajach jest związany z rezygnacją ze szczepienia przeciw gruźlicy[52][63]. Część autorów uważa nieszczepienie przeciw gruźlicy za czynnik ryzyka rozwoju czerniaka[66].
Epidemiologia
Czerniak stanowi 1% nowotworów złośliwych[29] i 5–7% nowotworów skóry u człowieka[67]. Na świecie pod względem zapadalności stanowi szesnasty najczęściej rozpoznawany nowotwór złośliwy u mężczyzn i piętnasty nowotwór złośliwy u kobiet[68]. Co roku na świecie diagnozuje się 132 000 przypadków czerniaka[69]. Zachorowalność na czerniaka systematycznie wzrasta, obserwuje się coroczny wzrost zapadalności na ten nowotwór o 3–7%[70][12][71][29][72]. Częściowo jest to związane z większą wykrywalnością i świadomością społeczną, ale również w związku ze zwiększoną ekspozycją na naturalne i sztuczne promieniowanie ultrafioletowe[12].
Częstość występowania choroby znacząco różni się pod względem geograficznym. Czerniak w 80% przypadków dotyczy białej populacji Europy, Ameryki Północnej i Australii[68]. Średnia zapadalność na świecie wynosi 4–12 na 100 000[73][74]. Najwyższą zapadalność obserwuje się w Australii (39/100 000), Nowej Zelandii (34/100 000), Stanach Zjednoczonych (17/100 000) i Skandynawii (12–15/100 000). W pozostałych krajach europejskich obserwuje się niższą zapadalność (4–10/100 000). W Afryce, Azji, Ameryce Południowej i Oceanii wśród populacji niebiałej obserwuje się niską zapadalność (3/100 000)[75].
Polska należy do krajów o niskiej częstości zachorowania na czerniaka[74]. Szacuje się, że w 2013 roku na czerniaka skóry zachorowało w Polsce 1400 mężczyzn i 1500 kobiet[76]. Zapadalność na czerniaka wynosi 4,5/100 000 u mężczyzn i 4,2/100 000 u kobiet[67]. Obserwuje się systematyczny wzrost zapadalności wynoszący 2,6% dla mężczyzn i 4,4% dla kobiet[74].
Czerniak przed 40. rokiem życia jest rzadki. Od 40. roku życia częstość zachorowania systematycznie rośnie i osiąga szczyt w siódmej i ósmej dekadzie życia[77]. Rozkład płci chorych jest różny w różnych populacjach. W populacjach o słabszym nasłonecznieniu częściej chorują kobiety[13]. W obszarach o dużej częstości występowania czerniaka istnieje tylko niewielka przewaga częstości zachorowania wśród kobiet lub zapadalność jest porównywalna pomiędzy płciami[78]. Istnieją różnice szczytu wieku zachorowania pomiędzy różnymi lokalizacjami anatomicznymi. Czerniak w obrębie tułowia osiąga szczyt zapadalności w piątej i szóstej dekadzie życia, a w obrębie głowy i szyi w ósmej dekadzie[13].
Histopatologia

.jpg)
_at_thigh_Case_01.jpg)


W klasyfikacji WHO do złośliwych nowotworów melanocytowych należą[79]:
- czerniak szerzący się powierzchownie (ang. superficial spreading melanoma, ICD-O 8743/3),
- czerniak guzkowy (ang. nodular melanoma, ICD-O 8721/3),
- czerniak z plamy soczewicowatej (ang. lentigo maligna, ICD-O 8742/2),
- czerniak podpaznokciowo-kończynowy (ang. acral-lentiginous melanoma, ICD-O 8744/3),
- czerniak desmoplastyczny (ang. desmoplastic melanoma, ICD-O 8745/3),
- czerniak wywodzący się ze znamienia błękitnego (ang. melanoma arising from blue naevus, ICD-O 8780/3),
- czerniak wywodzący się ze znamienia wrodzonego (ang. melanoma arising in a giant congenital naevus, ICD-O 8761/3),
- czerniak znamieniopodobny (ang. naevoid melanoma, ICD-O 8720/3).
Największe znaczenie kliniczne mają cztery typy czerniaka: czerniak szerzący się powierzchownie (ang. superficial spreading melanoma), czerniak guzkowy (ang. nodular melanoma), czerniak z plamy soczewicowatej (ang. lentigo maligna melanoma) i czerniak podpaznokciowo-kończynowy (dawniej postać akralna wywodząca się ze złośliwej plamy soczewicowatej – acral lentiginous melanoma)[52]. 80% wszystkich czerniaków u ludzi stanowią czerniak szerzący się powierzchownie i czerniak guzkowy[80].
Czerniak szerzący się powierzchownie
Jest to najczęstszy podtyp czerniaka stanowiący około 70% wszystkich przypadków tego nowotworu[81][82]. Charakteryzuje się promienistym i powierzchownym rozprzestrzenianiem się guza początkowo w naskórku, a w późniejszym etapie w warstwie brodawkowej i siateczkowej skóry właściwej. Czerniak szerzący się powierzchownie może pojawiać się w dowolnej lokalizacji, szczególnie w miejscach wystawionych na intensywną, przerywaną ekspozycję na promieniowanie ultrafioletowe. U kobiet zwykle występuje na kończynach, a u mężczyzn w obrębie tułowia[83].
Makroskopowo jest płaską lub płasko-wyniosłą zmianą z typową asymetrią jej budowy. Zmiana zwykle jest ostro ograniczona, a granica zmiany jest nieregularna, pokarbowana, może przypominać mapę[81][84]. Rzadziej granica jest nieostra[81]. Wykwit jest nierównomiernie zabarwiony, przybiera kolor od ciemnobrązowego do ciemnoczarnego. Szare i białe obszary wskazują na regresję. Czerwone obszary odpowiadają zapaleniu lub wzmożonemu unaczynieniu guza[81]. Możliwe jest występowanie bezbarwnych zmian, które przypominają chorobę Bowena lub Pageta[81].
W bardziej zaawansowanych stadiach czerniak szerzący się powierzchownie może osiągać znaczną średnicę i zmienia wzór wzrostu z horyzontalnego na pionowy, co klinicznie ujawnia się jako uniesienie zmiany. Powstają guzki łatwo ulegające erozji, owrzodzeniu i krwawiące. Możliwa jest obecność zmian satelitarnych[81].
Mikroskopowo czerniak szerzący się powierzchownie początkowo rozprzestrzenia się horyzontalnie (pagetoidalnie), głównie w naskórku powyżej połączenia skórno-naskórkowego, gdzie obecne są atypowe melanocyty. Nowotwór w późniejszym etapie wzrostu poziomego w wyniku nacieku również jest obecny w skórze właściwej w warstwie brodawkowatej i siateczkowatej. W czasie wzrostu poziomego w naskórku czerniak nie daje przerzutów i nie wykazuje oznak angiogenezy. Następnie dochodzi do wzrostu pionowego guza i nowotwór wrasta w głębsze warstwy skóry i zajmuje warstwy brodawkowatą i siateczkowatą[85][83].
Atypowe melanocyty są większe, mogą być wielokątnego lub owalnego kształtu, komórki zawierają obfitą ilość cytoplazmy, jądra posiadają nieregularnie grudkowaną chromatynę i cienką błonę jądrową[86][81]. Melanocyty występują pojedynczo lub w gniazdach[81]. Dystrybucja i kształt gniazd jest nieregularny, gniazda charakterystyczne dla czerniaka są duże i mają słabe odgraniczenie[86]. Gniazda mogą ulegać zlewaniu[86][81]. W czerniaku in situ reakcja zapalna jest mało nasilona lub nieobecna[81]. Widać liczne mitozy, figury mitotyczne mogą nie występować[86].
W fazie pionowego wzrostu występują obszerne i bardzo nieregularne gniazda atypowych melanocytów, które mogą znajdować się w znacznej odległości od siebie albo łączyć się. Warstwa naskórka jest nierównomiernie scieńczała, dochodzi do zajęcia przydatków skóry[86]. Komórki tracą cechy dojrzewania[81][85]. Mogą być obecne komórki, które uległy martwicy[86]. Obecny bywa naciek limfocytów[81].
Czerniak może ulegać częściowej lub (znacznie rzadziej) całkowitej regresji, co może być powodem trudności diagnostycznych ze znalezieniem guza pierwotnego. W części guza, która uległa regresji, ilość melanocytów jest znacznie zmniejszona w stosunku do pozostałej części zmiany. Dochodzi do zwłóknienia w warstwie brodawkowej, proliferacji naczyń i ich poszerzenia oraz różnego stopnia nacieku limfocytów i melanofagów[81].
Czerniak guzkowy
Jest to drugi co do częstości podtyp czerniaka, stanowi około 10–15% wszystkich przypadków czerniaka[87][82]. Charakteryzuje się pionowym wzorcem wzrostu. Czerniak guzkowy może występować w dowolnym miejscu, w porównaniu do czerniaka szerzącego się powierzchownie częściej występuje na tułowiu, głowie, szyi i kończynach dolnych[87]. Klinicznie jest szybko rosnącym guzem[88]. Czerniak guzkowy najczęściej powstaje de novo, a znacznie rzadziej w istniejącym wcześniej znamieniu[89]. Nowotwór ten statystycznie częściej niż czerniak szerzący się powierzchownie dotyka osoby starsze[87].
Makroskopowo przedstawia się jako szybko rosnąca grudka lub blaszka. Zmiana bywa polipowata, a rzadko nawet uszypułowa. Guz jest symetryczny i dobrze odgraniczony, o gładkiej powierzchni[88]. Zmiana cechuje się kolorem czarnym lub niebieskim, możliwa jest postać amelanocytowa, która przybiera kolor różowoczerwony[87]. Rozmieszczenie barwnika często nie jest symetryczne, choć bywa regularne[88]. Często jest obecne owrzodzenie guza[87][90].
Mikroskopowo jest podobny do czerniaka szerzącego się powierzchownie, jednak nie obserwuje się horyzontalnego śródnaskórkowego rozprzestrzeniania się. Komórki nowotworowe znajdują się w skórze właściwej, komponenta naskórkowa jest zależna od obecności czerniaka w skórze właściwej i nie istnieje samodzielnie[88]. Pokrywający naskórek jest scieńczały, zamazany lub owrzodziały[87]. Guz jest zbudowany z masywnych gniazd melanocytów[91]. Wyróżnia się typy o komórkach przypominających komórki nabłonkowe, typ o komórkach wrzecionowatych i typ mieszany[92][90]. Melanocyty zwykle są pleomorficzne i często stwierdza się kilka typów komórek[88]. Mitozy są liczne, często obecna jest martwica atypowych melanocytów[91].
Czerniak z plamy soczewicowatej


Czerniak z plamy soczewicowatej stanowi około 5% przypadków czerniaka[82]. Złośliwa plama soczewicowata jest uważana za czerniaka in situ, a czerniak z plamy soczewicowatej za jego inwazyjną formę. Czerniak z plamy soczewicowatej występuje u osób starszych. W przeciwieństwie do pozostałych form czerniaka etiologicznie wiąże się ze skumulowanym przewlekłym narażeniem na światło słoneczne i występujące w nim promieniowanie ultrafioletowe, co bardziej przypomina nieczerniakowe nowotwory skóry. Występuje na skórze uszkodzonej przez słońce, często jest poprzedzony wieloletnim występowaniem plamy soczewicowatej. Zwykle występuje w obrębie głowy i szyi, w pozostałych lokalizacjach obserwuje się go rzadziej[93]. Czerniak rozwija się po dłuższym, wieloletnim, przewlekłym przebiegu, przekraczającym 20 lat[29][94].
Makroskopowo są to obszerne, asymetryczne, płaskie zmiany o nieregularnej pigmentacji i słabym odgraniczeniu. Wraz ze wzrostem nasilają się różnice pigmentacji, mogą pojawiać się guzki w obrębie zmiany, a granica zmiany staje się trudna do określenia[95].
Złośliwa plama soczewicowata charakteryzuje się proliferacją atypowych melanocytów na granicy skórno-naskórkowej, często rozprzestrzeniających się w głąb wzdłuż mieszka włosowego lub gruczołu potowego, w niektórych obszarach atypowe melanocyty mogą być zlokalizowane poza warstwą podstawną. Melanocyty mogą układać się we wzór liniowy. Pojedyncze gniazda atypowych melanocytów są obecne na granicy skórno-naskórkowej. Melanocyty wykazują wyraźny pleomorfizm, widoczne jest zmniejszenie objętości cytoplazmy, jądro jest gwieździstego, półksiężycowatego lub okrągłego kształtu. Często obecny jest naciek limfocytarny[96].
W czerniaku z plamy soczewicowatej występują cechy inwazji skóry właściwej. Element naskórkowy jest podobny do złośliwej plamy soczewicowatej. Na element inwazyjny składają się okrągłe, nabłonkowate lub wrzecionowate komórki[94]. Atypowe komórki mogą występować w grupach lub w postaci pasm. Ze względu na szerzenie się wzdłuż gruczołów potowych i mieszka włosowego mogą występować trudności z oceną głębokości naciekania. Obecność bezbarwnych stref na brzegu zmiany może utrudniać ustalenie odpowiednio szerokiego marginesu zdrowych tkanek wokół usuwanej zmiany[96].
Czerniak podpaznokciowo-kończynowy


Jest to odrębna odmiana czerniaka występująca na dłoniach, stopach i podpaznokciowo z charakterystycznym obrazem histologicznym. Stanowi około 7–10% czerniaków[97][80][82]. Jednak jest znacznie częstszy u czarnoskórych i Azjatów, gdzie może być najczęstszą odmianą czerniaka w tych populacjach. Występuje głównie u starszych ludzi, częściej u mężczyzn[97]. Pojawia się w obrębie nieowłosionej skóry dłoni i stóp oraz podpoznokciowo. Niektóre definicje obejmują również grzbietową część dłoni. W 87% przypadków dotyczy stóp – w 57% jest zlokalizowany po stronie podeszwowej, 5% pod paznokciem i 9% po stronie grzbietowej. W 23% przypadkach pojawia się w obrębie dłoni – po stronie dłoniowej w 1%, grzbietowej w 9% i pod paznokciem w 14%[98]. Często dotyczy kciuka i dużego palca u stopy. Prawdopodobnie promieniowanie ultrafioletowe odgrywa w patogenezie czerniaka podpaznokciowo-kończynowego niewielką rolę[98].
Początkowo jest podobny do złośliwej plamy soczewicowatej. Wykazuje dwufazowy charakter wzrostu, początkowo o promienistym wzorcu wzrostu, następnie wzorzec pionowy. Klinicznie w fazie wzrostu poziomego występuje jako plamkowato nierównomiernie zabarwiona zmiana o nieregularnych, nierównych granicach. W fazie wzrostu pionowego w jej obrębie pojawiają się guzki. Częściej niż w innych podtypach występuje owrzodzenie[98].
Czerniak w obrębie paznokcia często zaczyna się jako plama pod paznokciem o odcieniu od brązowego do czarnego, często o smugowatej, pasmowatej pigmentacji. Dochodzi do pogrubienia, rozwarstwienia i zniszczenia płytki paznokciowej. Przebarwienie szerzy się z płytki paznokciowej na obrąbek naskórkowy lub opuszkę palca (objaw Hutchinsona)[29][98].
Mikroskopowo czerniak podpaznokciowo-kończynowy wykazuje charakterystyczny obraz histopatologiczny. W fazie promienistego wzrostu zmiana charakteryzuje się obecnością nadmiernego rogowacenia, poszerzenia warstwy rogowej naskórka, wydłużeniem listewek (sopli) naskórkowych i proliferacją atypowych melanocytów na granicy skórno-naskórkowej. Atypowe melanocyty są duże, zawierają powiększone jądra i dziwaczne jąderka, w cytoplazmie są obecne ziarnistości wypełnione melaniną. Rozprzestrzeniają się one w głąb skóry wzdłuż mieszka włosowego lub gruczołu potowego. W fazie pionowego wzrostu guz zawiera głównie komórki wrzecionowate, które są związane z reakcją desmoplastyczną (odczyn włóknisty)[99].
Czerniak desmoplastyczny

Czerniak desmoplastyczny stanowi około 1–4% czerniaków, częściej dotyczy mężczyzn niż kobiet. Większość zmian występuje w obrębie skóry wystawionej na działanie promieni słonecznych, choć może występować w miejscach osłoniętych. Najczęściej lokalizuje się w obrębie głowy i szyi, w tym ucha, nosa i wargi[100].
Makroskopowo występuje jako plamka, blaszka lub guzek. Często występuje niedobór lub brak pigmentacji. Mikroskopowo czerniak desmoplastyczny jest zbudowany z wrzecionowatych melanocytów przypominających fibroblasty, guzy zwykle są bezbarwne. Atypowe melanocyty układają się wśród wiązek kolagenu, związanych z zagęszczeniem i włóknieniem zrębu. Rozkład komórek jest bezładny, rzadziej tworzą one równoległe wiązki. Nowotwór może głęboko naciekać, sięgając do tkanki podskórnej oraz okostnej, szczególnie w obrębie czaszki[100].
Czerniak desmoplastyczny może wykazywać neurotropizm charakteryzujący się obecnością co najmniej jednego ogniska nacieku wrzecionowatych komórek wokół nerwu w skórze lub głębszych warstwach daleko od guza[101].
Czerniak wywodzący się ze znamienia błękitnego

Jest to bardzo rzadka odmiana związana ze skórną melanocytozą, najczęściej w związku ze znamieniem błękitnym[102]. Znamię błękitne jest to nagromadzenie w skórze melanocytów i melanoforów zabarwiających znamię na szaroniebiesko[103].
Czerniak wywodzący się ze znamienia błękitnego dotyczy osób stosunkowo młodych, średnia wieku zachorowania wynosi około 40 lat. Zwykle dotyczy osób rasy białej, nieco częściej mężczyzn. Najczęściej jest zlokalizowany w obrębie skalpu (33%), oczodołu i twarzy (32%), pleców i pośladków, kończyn, dłoni i stóp. Klinicznie stwierdza się niedawne szybkie powiększanie się znamienia błękitnego, zmiany koloru znamienia i owrzodzenie. Często jest to duży czarny guz ze zmianami satelitarnymi[102].
W obrazie mikroskopowym często jest widoczny łagodny histologicznie element znamienia błękitnego, który może dominować w obrazie histopatologicznym. Zwykle jest to połączenie komórkowego znamienia błękitnego i pospolitego znamienia błękitnego. Obszary złożone z komórkowego znamienia błękitnego są zbudowane z jajowatych, ściśle ułożonych komórek z obfitą cytoplazmą zawierającą niewielką ilość melaniny lub nieposiadającą jej w ogóle. Obszary złożone z pospolitego znamienia błękitnego są zbudowane z wrzecionowatych i dendrytycznych melanocytów obficie zawierających melaninę. Pomiędzy pęczkami dendrytycznych melanocytów widać melanofagi i wiązki kolagenu[104].
Komponent złośliwy zwykle występuje jako guzkowate skupiska atypowych melanocytów w warstwie siateczkowej skóry i tkance podskórnej. Atypowe melanocyty są duże i cechują się wrzecionowatym kształtem z obfitą cytoplazmą z licznymi figurami podziałowymi, z niewielką ilością lub bez obecności melaniny. Komórki są ułożone w warstwy, rozlanie naciekają głębokie warstwy skóry, niszcząc obecne tam struktury. Granica pomiędzy naciekanymi strukturami i czerniakiem jest ostra i poszarpana, często widać nagłe przejście z łagodnego znamienia błękitnego do czerniaka[104].
Czerniak wywodzący się z olbrzymiego znamienia wrodzonego

Olbrzymie wrodzone znamię występuje z częstością 1 na 20 000 niemowląt, a ryzyko transformacji nowotworowej do czerniaka jest szacowane na 6%. Olbrzymie znamię wrodzone zwykle jest definiowane jako znamię o wielkości przekraczającej 20 cm. Tułów i głowa są najczęstszymi lokalizacjami znamienia olbrzymiego, które może zajmować znaczny obszar, przekraczający 2% powierzchni ciała. Stosunkowo rzadko jest obecne od razu przy urodzeniu, częściej występuje jako szybko rosnąca plamka lub guzek o kolorze czarnoniebieskim, czerwonawym lub cielistym[105]. Większość czerniaków wywodzących się z olbrzymiego znamienia wrodzonego pojawia się przed 10. rokiem życia, drugi szczyt zachorowań występuje w życiu dorosłym. Tak jak znamię wrodzone, może występować w dowolnej lokalizacji, ale najczęściej jest obserwowany w obrębie tułowia[105].
Mikroskopowo jest dobrze odgraniczonym guzem w stosunku do olbrzymiego znamienia wrodzonego. Komórki nowotworowe często układają się w formie ekspansywnych guzków, a listewki naskórkowe są zamazane. Śródnaskórkowy element nowotworu jest utworzony przez melanocyty przypominające komórki nabłonkowe. Zmiana szerzy się pagetoidalnie (promieniście), często występuje owrzodzenie guza[106].
Czerniak znamieniopodobny
Stanowi około 1–2% czerniaków. Może wystąpić w każdym wieku, ale często dotyczy osób młodych i w średnim wieku. Zwykle jest zlokalizowany w obrębie tułowia, proksymalnych częściach kończyn oraz głowy i szyi[107].
Makroskopowo jest to niewielka grudka, guzek lub brodawkująca zmiana koloru od jasnobrązowego do ciemnobrązowego. Kolor może być jednolity lub nieregularny. Mikroskopowo cechuje się występowaniem obszarów warstwowej proliferacji atypowych melanocytów w skórze właściwej bez lub z niewielkim rozprzestrzenianiem się śródnaskórkowym[108].
Patogeneza
Powstanie czerniaka w wyniku transformacji złośliwej (karcynogenezy) melanocytów nie jest całkowicie poznane. U jej podstaw leży oddziaływanie szkodliwych czynników środowiskowych, które nakładają się na indywidualne cechy podatności i wywołują kaskadę kolejnych zmian genetycznych w melanocytach skutkujących ostatecznie wyselekcjonowaniem klonu nowotworowego[29]. Przejście prawidłowego melanocyta w komórkę nowotworową jest efektem zaburzenia regulacji w dół lub w górę wielu genów kontrolujących wzrost komórki, jej zdolności do podziału, unikania starzenia się komórki i ucieczki przed apoptozą (regulowanej śmierci komórki) oraz nasileniem angiogenezy[109]. Klonalne zaburzenia genetyczne są efektem mutacji genów delecji, amplifikacji, translokacji materiału genetycznego oraz zmian epigenetycznych polegających na stabilnym wyciszeniu transkrypcji genów bez fizycznej trwałej modyfikacji materiału genetycznego poprzez modyfikację chromatyny lub modyfikację histonów. W konsekwencji zmian powstaje klon atypowych melanocytów wykazujących przewagę w stosunku do otaczających ich komórek[109]. Tylko część mutacji daje przewagę proliferacyjną (podziałową) komórki nad prawidłowymi i sprzyja zwiększeniu potencjału komórki nowotworowej do przetrwania. Zmiany genetyczne nakładają się na siebie, tworząc liczne zróżnicowane klony, u których powtarzają się najwcześniejsze mutacje. Część z tych mutacji jest powtarzalna i ważna w patogenezie powstawania nowotworu – są to mutacje kierunkowe i mają ogromne znaczenie kliniczne, ponieważ stanowią potencjalny lub już wykorzystywany punkt uchwytu działania nowoczesnych celowanych leków przeciwnowotworowych[110][111][112][113].
Jednocześnie czerniak nie powstaje w jednorodnym mechanizmie patogenetycznym i nie jest jednorodną chorobą. Guzy różnią się pomiędzy sobą lokalizacją, posiadanymi mutacjami i ekspozycją na UV lub jej brakiem[112].
Kluczowa jest przemiana protoonkogenów do onkogenów, które nasilają wzrost komórki i zwiększają potencjał do podziałów (proliferację) mimo braku sygnałów wzrostowych ze strony środowiska zewnętrznego. W czerniaku zaobserwowano zaburzenia szlaku MAPK-ERK z kaskadą NRAS, BRAF, MEK1, MEK2, ERK1 i ERK2 wpływającym na powstanie czerniaka oraz na jego progresję. Szlak nasila wzrost atypowych melanocytów, zwiększa ich przeżywalność i oporność na apoptozę. Mniejsze znaczenie ma szlak RB[109][114][115][116].
Istotne jest unikanie starzenia się komórki rozumianego jako zatrzymanie proliferacji komórki w wyniku skrócenia telomerów lub stresu oksydacyjnego. W czerniaku stwierdzono zwiększenie aktywności telomerazy zapobiegającej skracaniu telomerów. Jednak komórki czerniaka mogą się dzielić w nieskończoność (tzw. nieśmiertelność komórki) mimo braku zwiększenia aktywności telomerazy. Mutacja genów supresorowych kontrolujących podziały komórkowe i stabilność genetyczną komórki pozwala uniknąć zablokowania nadmiernej proliferacji atypowych melanocytów oraz unikać apoptozy. Białko p16 wykazuje silny efekt antyproliferacyjny poprzez wiązanie kinaz CDK4 i CDK6 i zablokowanie fosforylacji RB1. Wykazano, że dysfunkcja szlaku p16 sprzyja niekontrolowanym podziałom komórek nowotworowych w czerniaku. Z kolei białko p14 wywiera silny efekt supresji nowotworu poprzez wiązanie białka MDM2, które blokuje p53 wykazujące silne zdolności do aktywacji naprawy DNA lub indukcji apoptozy komórki[109].
Historia naturalna choroby


Co najmniej połowa czerniaków pojawia się de novo na skórze niezmienionej bez poprzedzającego go znamienia barwnikowego, choć około 40% z nich rozwija się w obrębie znamiona barwnikowego[2]. Większość czerniaków początkowo rośnie jako płaska zmiana. Atypowe komórki rozprzestrzeniają się horyzontalnie wzdłuż zmiany (faza promienistego wzrostu), mogą być ograniczone do naskórka jako czerniak in situ lub być obecne w skórze właściwej jako czerniak inwazyjny, który nie posiada jeszcze zdolności do namnażania się w skórze właściwej[117]. Atypowe melanocyty mogą występować pojedynczo lub w gniazdach, a także migrować do warstwy brodawkowatej skóry, gdzie mogą ulegać apoptozie lub przetrwać i akumulować się[117]. W tym etapie nowotwór nie jest zdolny do wytwarzania przerzutów[34]. Kolejny etap charakteryzuje pionowy wzór wzrostu, w którym guz nacieka coraz głębsze warstwy skóry. Wówczas komórki nowotworowe już posiadają zdolność do przetrwania i namnażania się w skórze właściwej. Klinicznie przejawia się to jako powstanie guzka w obrębie płaskiej zmiany lub de novo jako postać guzkowa[117].
Większość przerzutów rozwija się w miarę postępu choroby, zwykle w ciągu kilku lat od wycięcia nowotworu. Rzadkie, ale możliwe jest ujawnienie się bardzo późnych przerzutów po ponad 10–25 latach od usunięcia guza pierwotnego[117]. Przerzuty odległe mogą powstawać z małych guzów[118]. Około 4% chorych jest diagnozowanych w stadium choroby przerzutowej[119]. Przerzuty rozprzestrzeniają się jednocześnie drogą układu limfatycznego oraz drogą krwionośną[118]. Zwykle przerzuty pojawiające się na drodze limfatycznej wyprzedzają przerzuty szerzące się drogą krwionośną[120]. Najczęściej rozwijają się one w trzech etapach. Początkowo są to przerzuty satelitarne i in transit (pomiędzy guzem pierwotnym a węzłem chłonnym), następnie przerzuty węzłowe i ostatecznie przerzuty odległe. Przerzuty odległe mogą być obecne bez zajęcia lokalnych węzłów chłonnych[117]. U około 50% chorych pierwszymi przerzutami są lokalne przerzuty węzłowe, u około 20% są to przerzuty in transit i satelitarne, a u 30% są od razu przerzuty odległe[118][121].
Przerzuty mogą występować w każdej lokalizacji, początkowo zwykle dotyczą skóry, tkanki podskórnej oraz węzłów chłonnych (42–60%). Trzewia są pierwszą lokalizacją przerzutów u 25% chorych. Najczęstszymi zajętymi narządami są płuca (18–36%), mózg (12–20%), wątroba (14–20%) i kości (11–17%)[119]. Lokalizacja przerzutów odległych jest jednym z najważniejszych czynników rokowniczych u chorych z chorobą przerzutową. Przeżycie chorych jest dłuższe w przypadku lokalizacji przerzutów w odległych węzłach chłonnych tkanek miękkich i skóry niż u chorych z przerzutami w trzewiach. Z kolei przerzuty w płucach rokują lepiej niż przerzuty w pozostałych lokalizacjach trzewnych[119]. Rzadko stwierdza się obecność przerzutów odległych bez widocznego guza pierwotnego, w niektórych przypadkach może być to spowodowane regresją guza pierwotnego lub ukrytą lokalizacją czerniaka w obrębie gałki ocznej lub błon śluzowych[122].
Rozpoznanie

Rozpoznanie kliniczne jest oparte o dermatoskopię, która polega na ocenie podejrzanych zmian i na podstawie odpowiednich kryteriów rozpoznania, wyznaczeniu zmian budzących podejrzenie nowotworu złośliwego do biopsji wycinającej. Pozostałe metody mają zastosowanie pomocnicze, są przydatne w przypadkach wątpliwych i trudnych diagnostycznie, kiedy nie można wykonać biopsji wycinającej. Podejrzana zmiana jest wycinana z niewielkim (1–2 mm) marginesem zdrowych tkanek. Ostateczne rozpoznanie stawia się na podstawie badania histopatologicznego, które pozwala również ocenić zaawansowanie nowotworu, wolny od nowotworu margines resekcji oraz inne czynniki ważne dla rokowania i leczenia.
Dermatoskopia
Dermatoskopia (dermoskopia, mikroskopia epiluminescencyjna) jest to nieinwazyjna metoda badania skóry pozwalająca na ocenę naskórka i skóry właściwej. Dermatoskopy oferują powiększenie od 10-krotnego w tradycyjnych dermatoskopach do 100-krotnego w wideodermatoskopach. Pozwala to na lepsze zobrazowanie i łatwiejszą ocenę zmian barwnikowych niż w przypadku nieuzbrojonego oka. Kluczowa jest ocena wszystkich zmian skórnych[123]. Dermatoskopy mogą używać źródła światła spolaryzowanego lub niespolaryzowanego, które wymaga immersji, zwykle za pomocą odpowiedniego olejka, wody lub żelu ultrasonograficznego[123]. Znamiona melanocytowe podczas niektórych stanów (ciąża, dzieciństwo, intensywna ekspozycja na promieniowanie ultrafioletowe) mogą przybierać niezwykłe formy dermatoskopowe. Część autorów nie zaleca rutynowego badania dermatoskopowego w ciągu 2 miesięcy od intensywnej ekspozycji na promieniowanie ultrafioletowe[124].
Badanie rozpoczyna się od obejrzenia wszystkich zmian skórnych pod dobrym oświetleniem bez użycia dermatoskopu (badanie „nieuzbrojonym okiem”). Następnie ocenia się za pomocą dermatoskopu możliwie wszystkie zmiany skórne, szczególnie te podlegające zmianom koloru, wielkości, kształtu, powierzchni, dające objawy takie jak świąd, pieczenie, krwawienie, stan zapalny, a także zmiany różniące się od pozostałych znamion (objaw „ugly duckling”)[123]. Badanie uwzględnia również specjalne okolice obejmujące owłosioną część głowy, dłonie, stopy, przestrzenie międzypalcowe i dostępne w badaniu błony śluzowe[124].
Kolejnym etapem jest odróżnienie zmian niemelanocytowych od melanocytowych. Następnie zmiany melanocytowe ocenia się według jednego z kilku systemów oceny, do których należą: reguła trzypunktowa, dermatoskopowa reguła ABCD, reguła siedmiopunktowa i metoda Menziesa. Ocena zmian uwzględnia kliniczny przebieg oceniany na podstawie wywiadu lub dokumentacji fotograficznej. Na podstawie kryteriów poszczególnych metod ustala się podejrzenie kliniczne czerniaka[125]. W przypadkach zmian wątpliwych w dermatoskopii, kiedy nie można wykluczyć nowotworu złośliwego, zmiany kwalifikuje się do wycięcia, co wynika z założenia, że nawet niepotrzebna procedura chirurgiczna jest bardziej korzystna niż ryzyko przeoczenia nowotworu złośliwego[125][123].
Ocena cyfrowa obrazów wideodermatoskopowych może być przydatna w diagnostyce czerniaka. Obrazy te wykazują wyższą czułość, ale niższą swoistość niż badanie dermatoskopowe wykonywane przez doświadczonego lekarza[126].
Dermatoskopia w wykonaniu doświadczonych w badaniu lekarzy pozwala osiągnąć czułość 81,3% i swoistość 94,6%[126]. Metoda umożliwia wykrycie wczesnego czerniaka oraz jednocześnie pozwala zredukować ilość łagodnych zmian kwalifikowanych do nieprzynoszącego korzyści chirurgicznego wycięcia[127][124][128].
Wszystkie zmiany odróżniające się od pozostałych oraz zmiany budzące wątpliwość badającego lekarza są kwalifikowane do usunięcia, a właściwie do biopsji wycinającej i następnie badania histopatologicznego[123]. U chorych ze znaczną ilością znamion melanocytowych zaleca się regularne kontrole, które pozwalają odpowiednio wcześnie wykryć czerniaka wśród licznych podobnych zmian, a jednocześnie zapobiec nadmiernej liczbie interwencji chirurgicznych[123]. U osób dorosłych znamiona błękitne o nieustalonym wywiadzie lub o zmieniającym się obrazie dermatoskopowym oraz znamiona Spitz, w związku z dużymi trudnościami w odróżnieniu tych zmian od czerniaka, kwalifikuje się do profilaktycznego wycięcia niezależnie od klinicznego podejrzenia nowotworu złośliwego[123]. Szybkie powiększanie się zmiany na dłoniach i stopach mimo łagodnego obrazu dermatoskopowego również może wskazywać na czerniaka i kwalifikuje się do wycięcia[125].
Kwalifikują się do chirurgicznego wycięcia[123]:
- zmiany, w których dermatoskopowo stwierdzono istotne różnice w porównaniu do poprzednich badań,
- zmiany z ekcentrycznym przebarwieniem, gdy są obecne inne struktury,
- zmiany o charakterze guzkowym,
- zmiany o szarawym lub białawym zabarwieniu związanym z regresją, szczególnie gdy przekracza ona 10% powierzchni,
- zmiany z obwódką, które powiększyły swoje rozmiary,
- zmiany z obecnością obwodowych ciałek barwnikowych po 60. roku życia,
- zmiany typu „wybuch gwiazdy” – promieniste wypustki wokół zwykle homogennego centrum,
- znamiona błękitne, jeśli czas ich powstania jest nieznany lub dochodzi do zmian w obrębie znamienia,
- zmiany uprzednio leczone za pomocą kriochirurgii, laseroterapii i innych metod niepozwalających na ocenę histopatologiczną całej zmiany,
- zmiany bezbarwnikowe w obrębie, których stwierdza się mleczno-czerwone obszary bezstrukturalne lub atypowe naczynia,
- zmiany „spitzoidalne”.
| Kryteria | Punkty |
| asymetria koloru lub struktury | 1 |
| atypowa siatka bawnikowa | 1 |
| obecność niebiesko-białych struktur welonowatych | 1 |
| Kryterium rozpoznania czerniaka | |
| ≥2 punkty wskazują na duże ryzyko występowania czerniaka | |
| Kryteria większe | Punkty |
| atypowa siatka barwnikowa (brązowa, czarna, szara, nieregularna) | 2 |
| objaw welonu | 2 |
| nieprawidłowy wzorzec naczyniowy (naczynia nieregularne linijne i typu kropek) | 2 |
| Kryteria mniejsze | Punkty |
| nieregularne wypustki, smugi | 1 |
| nieregularne kropki i ciałka skupione | 1 |
| nieregularne plamy | 1 |
| struktury regresyjne | 1 |
| Kryterium rozpoznania czerniaka | |
| Czerniaka rozpoznaje się gdy zmiana osiąga >3 punktów | |
| Cecha | Opis | Punktacja |
| A – (asymetry) asymetria | Ocena względem dwóch osi asymetrii kształtu, wybarwienia i struktur | 0-2 * 1,3 |
| B – (border) odgraniczenie od zdrowej skóry | Nagłe zakończenie wzoru zabarwienia w częściach obwodowych w 8 segmentach | 0-8 * 0,1 |
| C – (color) barwa | Punkt za obecność każdej z barw: jasnobrązowa, ciemnobrązowa, czarna, biała, czerwona, szaroniebieska | 0-6 * 0,5 |
| D – (different) obecność struktur różnicujących | Punkt za obecność każdej ze struktur: siatka barwnikowa, smugi gałązkowate, ciałka skupione, kropki barwnikowe, obszary bezstrukturalne | 1-5 * 0,5 |
| Interpretacja | ||
| 1,0-4,75 – zmiana łagodna | ||
| 4,8-5,45 – zmiana podejrzana (obserwacja lub profilaktyczne wycięcie) | ||
| 5,46-8,9 – wysoce podejrzana | ||
| Cechy wykluczające | Cechy potwierdzające |
| Symetria wzorca barwnikowego Obecność wyłącznie jednego koloru zmiany | Objaw niebiesko-białego welonu Liczne brązowe kropki Pseudowypustki Promieniście rozchodzące się smugi (ang. radial streaming) Białe obszary odbarwienia przypominające bliznę Obwodowe czarne kropki i ciałka skupione Wielobarwność zmiany (5-6 kolorów) Liczne niebiesko-szare kropki przypominające ziarna pieprzu Poszerzona siatka barwnikowa |
| Kryterium rozpoznania czerniaka | |
| Musi być spełniona co najmniej jedna z cech potwierdzających i równocześnie nie może pojawiać się żadna z cech wykluczających | |
Refleksyjna mikroskopia konfokalna
Refleksyjna mikroskopia konfokalna (reflectance mode confocal microscopy, RCM) jest to nieinwazyjna metoda diagnostyczna umożliwiająca zobrazowanie z dużą dokładnością naskórka i górnej części skóry właściwej. Odmianą badania jest laserowa refleksyjna mikroskopia konfokalna. Metoda jest oparta o różnice współczynnika odbicia wiązki promienia świetlnego przez różne obiekty, co pozwala odwzorować jej strukturę. Refleksyjna mikroskopia konfokalna jest szczególnie przydatna w różnicowaniu czerniaka bezbarwnikowego (amelanocytarnego)[123].
Rozpoznanie czerniaka jest oparte na stwierdzeniu nieregularności kształtu i rozmieszczenia, nieostro zarysowanych brodawek skórnych, łagodnej lub zaznaczonej na połączeniu skórno-naskórkowym atypii komórkowej (główne kryteria), a także obecności komórek pagetoidalnych (duże, okrągłe komórki z załamującą światło cytoplazmą i ciemnym jądrem) w bardziej powierzchownej warstwie naskórka lub nacieku pagetoidalnego, klastrów komórek przypominających kształtem mózg w warstwie brodawkowatej i obecności jądrzastych komórek w brodawkach skórnych (kryteria mniejsze). Do rozpoznania czerniaka konieczne jest spełnienie jednego kryterium głównego i jednego mniejszego[123]. Przy tych kryteriach badanie pozwala osiągnąć 90% czułości i 70% swoistości w wykrywaniu czerniaka[123]. Owrzodzenie lub nadmierne rogowacenie zmiany może utrudniać badanie i wówczas może być wskazana biopsja wycinająca[130].
Metoda znajduje zastosowanie w przypadkach wątpliwych, gdy wycięcie podejrzanej zmiany jest niemożliwe lub bardzo trudne (rozległe znamiona wrodzone)[3].
Ultrasonografia wysokiej częstotliwości
Standardowe przetworniki aparatów ultrasonograficznych o częstotliwości 7,5–15 MHz nie pozwalają na zobrazowanie struktur mniejszych od 1 mm. Zastosowanie głowic z przetwornikami o częstotliwości 20–100 MHz umożliwia obrazowanie skóry[131].
W USG czerniak jest zmianą hipoechogenną, dobrze odgraniczoną od sąsiednich struktur i bogato unaczynioną. Naciek zapalny towarzyszący guzowi może być trudny do odróżnienia od czerniaka i może powodować przeszacowanie jego grubości. Z kolei hiperkeratoza powoduje niedoszacowanie grubości, dlatego dokładność badania jest ograniczona w zmianach na dłoniach i stopach, gdzie występuje bardzo gruba warstwa naskórka[131].
Ultrasonografia wysokiej częstotliwości uzupełnia klasyczne metody diagnostyczne[131]. Jest przydatnym badaniem pozwalającym ocenić grubość czerniaka, unaczynienie nowotworu i potencjał do przerzutowania, co pomaga odpowiednio zaplanować leczenie[131][132].
Koherencyjna tomografia optyczna
Koherencyjna tomografia optyczna to nieinwazyjna technika obrazowania wykorzystująca promieniowanie podczerwone i znajdująca zastosowanie przede wszystkim w okulistyce. Umożliwia ona uwidocznienie struktur skóry wielkości 3-15 µm na głębokości do około 1 mm. Metoda pozwala również na badanie zmian hiperkeratotycznych, a także zmian na dłoniach i stopach. Jej ograniczeniem jest niemożliwość zbadania zmian większych od 1 mm[124].
Biopsja wycinająca

Biopsja wycinająca całą zmianę jest postępowaniem z wyboru w przypadku klinicznego podejrzenia czerniaka. Zabieg ma na celu dostarczenie próbki do badania histopatologicznego i ostatecznego rozpoznania czerniaka lub ewentualnego jego wykluczenia. Nie ma wskazań do profilaktycznego wycinania znamion niebudzących podejrzeń czerniaka lub innego nowotworu złośliwego[3].
Wycina się całą zmianę z wąskim (1–2 mm) marginesem prawidłowej skóry, wycięcie obejmuje całą grubość skóry i sięga do tkanki podskórnej, która nie musi zostać całkowicie wycięta, z wyjątkiem bardzo szczupłych chorych[133]. Biopsja jest tak planowa, aby w przypadku rozpoznania nowotworu złośliwego nie utrudniała późniejszego leczenia chirurgicznego oraz ewentualnej biopsji węzła wartowniczego. W związku z tym unika się szerokich marginesów podczas diagnostycznego wycięcia zmiany i poprzecznej linii wycięcia zmian na kończynach[134].
Biopsja wycinająca podejrzane zmiany może być nieakceptowalna w przypadku zmian na twarzy, genitaliów, powierzchni dłoniowej ręki, podeszwowej stóp, opuszek palców i okolicy paznokciowej, a także bardzo rozległych zmian. Wówczas można zastosować biopsję punktową (punch biopsy) lub biopsję nacinającą pełną grubość skóry (bez wycięcia całej zmiany)[133][134]. Takie biopsje powinny zawierać część krawędzi zmiany, której ocena jest istotnym elementem badania histopatologicznego[134].
W przypadku, gdy wstępna biopsja nie pozwala na wykluczenie czerniaka lub nie jest możliwa ocena lokalnego zaawansowania klinicznego, zaleca się rozważyć ponowną biopsję wycinającą[134]. Nie zaleca się biopsji ścinającej, ponieważ bywa przyczyną przeoczenia nowotworu złośliwego z powodu niemożliwości oceny całej zmiany[133].
Trudności diagnostycznych dostarcza lokalizacja w okolicy podpaznokciowej (czerniak podpaznokciowo-kończynowy). Zmiana często jest rozciągnięta na całej długości paznokcia i często rozpoczyna się u podstawy paznokcia. Biopsja wycinająca wymaga usunięcia znacznej części paznokcia. Alternatywą jest kilka biopsji punktowych, choć może okazać się konieczne kilkakrotne powtarzanie procedury[133].
Badanie histopatologiczne
.jpg)
Badanie histopatologiczne materiału uzyskane w wyniku biopsji podejrzanej klinicznie zmiany jest podstawą do ostatecznego rozpoznania czerniaka, a także do rozpoznania podtypu histologicznego[4].
Poza ostatecznym rozpoznaniem choroby nowotworowej badanie to dostarcza cennych informacji o znaczeniu rokowniczym i pozwala ocenić zaawansowanie kliniczne choroby. Badanie ocenia głębokość nacieku w skali Breslowa i stopień zajęcia warstw skóry według Clarka, margines wycięcia zmiany (radykalność), obecność zmian satelitarnych, obecność nacieku naczyń krwionośnych, naczyń limfatycznych, naciekanie nerwów (neurotropizm), obecność martwicy, owrzodzenia i indeks mitotyczny. Umożliwia rozpoznanie zajęcia lokalnych węzłów chłonnych i przerzutów odległych w bioptatach z podejrzanych klinicznie zmian[135].
Badania obrazowe

Badania radiologiczne znajdują zastosowanie przede wszystkim w ocenie zaawansowania klinicznego i wykazują niewielką przydatność w pierwotnej diagnostyce czerniaka.
- Tomografia komputerowa (TK)
Tomografia komputerowa jest najbardziej czułą metodą w wykrywaniu przerzutów w płucach (jednak do rutynowej oceny zaawansowania zaleca się RTG klatki piersiowej). Jest przydatną metodą w wykrywaniu przerzutów w kościach, szczególnie zmian litycznych (niszczących kość) niewidocznych w scyntygrafii. Tomografia jest wykonywana celem oceny zaawansowania choroby w obrębie jamy brzusznej, gdzie tomografia osiąga podobną czułość, ale wyższą swoistość diagnostyczną niż USG[136].
- Rezonans magnetyczny (MRI)
Rezonans magnetyczny jest bardziej czułą metodą diagnostyczną niż tomografia komputerowa w wykrywaniu przerzutów w mózgu, rdzeniu kręgowym i oponach mózgowo-rdzeniowych. Jest to najlepsza metoda stwierdzająca patologiczną masę w szpiku kostnym. Rezonans magnetyczny pozwala na różnicowanie łagodnych nowotworów wątroby, w tym z naczyniakami, od nowotworu przerzutowego[136].
- Pozytonowa tomografia emisyjna (PET)
Pozytonowa tomografia emisyjna szczególnie w połączeniu z tomografią komputerową (PET-TK) stanowi cenne narzędzie w wykrywaniu przerzutów. Jest to czulsza metoda w wykrywaniu przerzutów odległych od tomografii komputerowej czy rezonansu magnetycznego i jest co najmniej tak samo swoista jak te metody. Czułość w wykrywaniu przerzutów >1 cm wynosi powyżej 90%[136][137][138][139]. Ponadto metoda może skutecznie uwidaczniać przerzuty mniejszej wielkości, jeśli tło tkankowe jest niskiej aktywności metabolicznej[136].
PET-TK jest bardziej czułą metodą niż PET w wykrywaniu przerzutów w mózgu, wątrobie, węzłach chłonnych[137][140], a także kościach[141][142]. Jednocześnie jednak nie ma dowodów na korzyści wynikające z wczesnego wykrycia przerzutów[136].
- Scyntygrafia kości
Scyntygrafia pozwala na wcześniejsze wykrycie przerzutów niż zdjęcie RTG. Jednak scyntygrafia jest wykonywana wyłącznie u chorych z obrazem klinicznym wskazującym na obecność przerzutów kostnych[136].
Ocena rokowania
Samo stwierdzenie obecności choroby nowotworowej jest niewystarczające do podjęcia decyzji o najkorzystniejszej i najskuteczniejszej metodzie leczenia. Konieczna jest ocena zaawansowania choroby, która pozwala dostosować strategię leczenia do obecnego stanu.
Czynniki rokownicze
| Stopień | Głębokość naciekania |
| I | Atypowe melanocyty (komórki nowotworowe) obecne w naskórku |
| II | Obecność atypowych melanocytów w warstwie brodawkowatej skóry |
| III | Obecność atypowych melanocytów w obrębie połączenia warstwy brodawkowatej i siateczkowatej |
| IV | Obecność atypowych melanocytów w warstwie siateczkowatej |
| V | Obecność atypowych melanocytów w tkance podskórnej |
Rokowanie jest uzależnione od wielu czynników i zależy od podtypu histopatologicznego czerniaka, jego lokalizacji, głębokości nacieku według skali Breslowa, stopnia zajęcia warstw skóry według Clarka i obecności owrzodzenia zmiany. Rokowanie jest generalnie gorsze u osób starszych niż młodych, oraz u mężczyzn w porównaniu do kobiet. Wykazano, że kobiety w nieprzerzutowym czerniaku wykazują znacząco dłuższe przeżycie dziesięcioletnie w porównaniu do mężczyzn[144].
Najsilniejszymi czynnikami rokowniczymi są owrzodzenie guza i jego grubość wyrażone w skali Breslowa (obecnie element klasyfikacji TNM) lub Clarka[145]. Ryzyko owrzodzenia zmiany rośnie wraz z grubością guza. Z kolei obecność owrzodzenia znacznie obniża odsetek przeżycia pięcioletniego we wszystkich kategoriach grubości guza w porównaniu do guzów bez owrzodzenia[146][147]. Również odsetek przeżyć pięcioletnich spada wraz z grubością guza[146].
Czerniak powierzchownie się szerzący rokuje lepiej niż inne typy histologiczne, ponieważ cechuje go niższa głębokość nacieku według Breslowa. Czerniak zlokalizowany w obrębie kończyn rokuje lepiej niż te w obrębie tułowia, głowy i szyi, które wiążą się z gorszym przeżyciem całkowitym[144][146][148].
| Stadium | Grubość |
| I | <0,75 mm |
| II | 0,76–1,50 mm |
| III | 1,51–2,25 mm |
| IV | 2,26–3,00 mm |
| V | >3,00 mm |
Wysoki indeks mitotyczny jest markerem złego rokowania. U chorych z wysokim indeksem mitotycznym szybciej dochodzi do nacieku naczyń krwionośnych i limfatycznych, zwiększając ryzyko zajęcia węzłów chłonnych[151][152]. Naciek limfocytarny nowotworu jest niekorzystnym czynnikiem rokowniczym o niewiele niższej sile jak indeks mitotyczny[147][146].
Rokowanie chorych jest gorsze w przypadku zajęcia węzłów chłonnych. Liczba zajętych węzłów chłonnych (cecha N) jest skorelowana z pięcioletnim przeżyciem, które jest mniejsze im więcej węzłów chłonnych jest zajętych przez przerzuty węzłowe[146]. Rokowanie jest także lepsze w przypadku nieobecności przerzutów odległych[146]. Ryzyko wystąpienia przerzutów węzłowych zwiększa się wraz z głębokością nacieku guza w skali Breslowa, indeksem mitotycznym, młodszym wiekiem chorego i zajęciem struktur naczyniowo-nerwowych[152][153][154]. Chorzy z indeksem mitotycznym powyżej 6 mają 12-krotnie zwiększone ryzyko wystąpienia przerzutów odległych[155].
W chorobie uogólnionej najważniejszym czynnikiem rokowniczym jest lokalizacja przerzutów. Lepiej rokują przerzuty nienarządowe niż narządowe[145].
Regresja czerniaka to degeneracja nowotworowych melanocytów i zastąpienie tkanki nowotworowej zwłóknieniem i naciekiem limfocytarnym oraz wytworzeniem teleangiektazji[146]. Częstość występowania regresji czerniaka o grubości poniżej 0,75 mm wynosi około 60%[156]. Większość badań wskazuje na jego niewielką rolę w prognozowaniu przebiegu choroby[146].
Klasyfikacja TNM
Klasyfikacja TNM jest systemem oceny zaawansowania klinicznego nowotworu.
| Guz pierwotny – cecha T | |
| Tx | Nie można ocenić guza pierwotnego |
| T0 | Nie stwierdza się guza pierwotnego |
| Tis | Czerniak in situ |
| T1 | Czerniak o grubości 1 mm lub mniejszej |
| T1a | Czerniak o grubości 1 mm lub mniejszej bez owrzodzenia i indeks mitotyczny <1/mm² |
| T1b | Czerniak o grubości 1 mm lub mniejszej z owrzodzeniem lub indeks mitotyczny >1/mm² |
| T2 | Czerniak o grubości 1,01–2,00 mm |
| T2a | Czerniak o grubości 1,01–2,00 mm bez owrzodzenia |
| T2b | Czerniak o grubości 1,01–2,00 mm z owrzodzeniem |
| T3 | Czerniak o grubości 2,01–4,00 mm |
| T3a | Czerniak o grubości 2,01–4,00 mm bez owrzodzenia |
| T3b | Czerniak o grubości 2,01–4,00 mm z owrzodzeniem |
| T4 | Czerniak o grubości powyżej 4,00 mm |
| T4a | Czerniak o grubości powyżej 4,00 mm bez owrzodzenia |
| T4b | Czerniak o grubości powyżej 4,00 mm z owrzodzeniem |
| Zajęcie okolicznych węzłów chłonnych – cecha N | |
| Nx | Nie można ocenić okolicznych węzłów chłonnych |
| N0 | Nie stwierdza się przerzutów w okolicznych węzłach chłonnych |
| N1 | Zajęcie 1 węzła chłonnego |
| N1a | Zajęcie 1 węzła chłonnego – mikroprzerzut |
| N1b | Zajęcie 1 węzła chłonnego – makroprzerzut |
| N2 | Zajęcie 2-3 węzłów chłonnych |
| N2a | Zajęcie 2-3 węzłów chłonnych – mikroprzerzut |
| N2b | Zajęcie 2-3 węzłów chłonnych – ≥1 makroprzerzut |
| N2c | Zajęcie 2-3 węzłów chłonnych – guzek satelitarny lub przerzut in transit bez zajęcia regionalnych węzłów chłonnych |
| N3 | Zajęcie >3 węzłów chłonnych, pakiet węzłów chłonnych; guzek satelitarny lub przerzut in transit z przerzutami w węzłach chłonnych |
| Przerzuty odległe – cecha M | |
| Mx | Nie można określić obecności przerzutów odległych |
| M0 | Nie stwierdza się przerzutów odległych |
| M1a | Obecne przerzuty odległe w skórze, tkance podskórnej lub odległych węzłach chłonnych przy prawidłowym stężeniu LDH |
| M1b | Obecne przerzuty odległe w płucach przy prawidłowym stężeniu LDH |
| M1c | Przerzuty w pozostałych narządach trzewnych przy prawidłowym stężeniu LDH w surowicy lub przerzuty odległe w dowolnej lokalizacji ze zwiększonym stężeniem LDH |
Kliniczna ocena stopnia zaawansowania nowotworu w klasyfikacji TNM obejmuje ocenę mikroskopową zaawansowania pierwotnego czerniaka, a także kliniczną i radiologiczną ocenę występowania przerzutów. Ocena patologiczna obejmuje dodatkowo mikroskopową ocenę zaawansowania pierwotnego guza oraz badanie regionalnych węzłów chłonnych[157][158].
Mikroprzerzuty są rozpoznawane na podstawie badania histopatologicznego materiału z biopsji węzła wartowniczego i limfadektomii. Makroprzerzuty są definiowane jako przerzuty stwierdzane klinicznie i potwierdzone badaniem histopatologicznym oraz w przypadku nacieku nowotworowego zajętego węzła przekraczającego jego torebkę[159][158].
Przerzuty w regionalnych węzłach chłonnych są klasyfikowane następująco:
- guzki satelitarne są definiowane jako widoczne makroskopowo przerzuty skórne w odległości poniżej 20 mm od ogniska pierwotnego.
- guzki mikrosatelitarne są definiowane jako ogniska nowotworu (gniazda atypowych melanocytów) o nieciągłym charakterze o średnicy przekraczającej 0,05 mm sąsiadującym z pierwotnym guzem w odległości przynajmniej 0,3 mm i oddzielonym od niego przez prawidłową skórę.
- przerzuty in transit są to przerzuty występujące powyżej 20 mm od ogniska pierwotnego w obszarze pomiędzy guzem pierwotnym i najbliższą grupą regionalnych węzłów chłonnych[158].
| Stopień zaawansowania | Cecha T | Cecha N | Cecha M |
| 0 | Tis | N0 | M0 |
| IA | T1a | N0 | M0 |
| IB | T1b | N0 | M0 |
| T2a | T0 | M0 | |
| IIA | T2b | N0 | M0 |
| T3a | N0 | M0 | |
| IIB | T3b | N0 | M0 |
| T4a | N0 | M0 | |
| IIC | T4b | N0 | M0 |
| III | dowolne T | ≥N1 | M0 |
| IV | dowolne T | dowolne N | M1 |
| Stopień zaawansowania | Cecha T | Cecha N | Cecha M |
| 0 | Tis | T0 | M0 |
| IA | T1a | T0 | M0 |
| IB | T1b | N0 | M0 |
| T2a | N1 | M0 | |
| IIA | T2b | N0 | M0 |
| T3a | N1 | M0 | |
| IIB | T3b | N0 | M0 |
| T4a | N1 | M0 | |
| IIC | T4b | N0 | M0 |
| IIIA | T1-4a | N1a | M0 |
| T1-4a | N2a | M0 | |
| IIIB | T1-4b | N1a | M0 |
| T1-4b | N2a | M0 | |
| T1-4a | N1b | M0 | |
| T1-4a | N2b | M0 | |
| T1-4a | N2c | M0 | |
| IIIC | T1-4b | N1b | M0 |
| T1-4b | N2b | M0 | |
| T1-4b | N1c | M0 | |
| dowolne T | N3 | M0 | |
| IV | dowolne T | dowolne N | M1 |
Leczenie
Podstawową metodą leczenia czerniaka jest wycięcie guza w zakresie odpowiednio szerokiego marginesu zdrowych tkanek. W guzach nieprzekraczających 1 mm konieczny jest margines o szerokości 1 cm, a szersze wycięcie przeprowadza się w czerniakach o grubości 1,01–2 mm, które wymagają 2 cm marginesu zdrowych tkanek. W stadium klinicznym IA i IB z guzem poniżej 0,75 mm, ze względu na niskie ryzyko przerzutów do węzłów chłonnych, nie zaleca się wykonywania biopsji węzła wartowniczego, jednak jest ona przeprowadzana w guzach o grubości powyżej 0,75–1,0 mm. Biopsje węzła wartowniczego wykonuje się u chorych w stadium IB, stadium II w guzach o grubości powyżej 0,75–1,0 mm oraz w stadium III. Węzeł wartowniczy jest oznaczany za pomocą limfoscyntygrafii lub metody barwnikowej. U wszystkich chorych w stadium klinicznym III oraz w przypadku zajęcia węzła wartowniczego wycina się węzły chłonne w dorzeczu spływu limfatycznego guza (limfadenektomia regionalna). Leczenie adiuwantowe za pomocą interferonu α przeprowadza się u niektórych chorych w stadium IIB, IIC oraz III. Choroba nieoperacyjna lub obecność przerzutów odległych wymaga leczenia ogólnoustrojowego, w którym wykorzystuje się immunoterapię, leczenie celowane i klasyczną chemioterapię. W terapii celowanej stosuje się inhibitory BRAF (wemurafenib, dabrafenib), inhibitory MEK (trametynib, selumetynib), inhibitory c-KIT (imatynib). Z kolei w immunoterapii wykorzystuje się przeciwciała anty-CTLA 4 (ipilimumab), przeciwciała anty-PD1 (pembrolizumab, niwolumab) oraz IL-2. Wybór najlepszej metody leczenia jest dostosowany indywidualnie do każdego pacjenta w zależności od obecnych mutacji oraz dotychczasowego przebiegu choroby. W leczeniu II linii stosuje się klasyczną chemioterapię lub biochemioterapię.
Leczenie chirurgiczne
Szerokie wycięcie czerniaka
Chirurgiczna szeroka resekcja czerniaka z odpowiednio szerokimi marginesami zdrowych tkanek stanowi podstawową metodę leczenia choroby zlokalizowanej (bez przerzutów)[160]. W większości przypadków jest to usunięcie blizny po wykonanej biopsji wycinającej. Umożliwia zabezpieczenie chorego przed wznową miejscową, a w chorobie bez przerzutów pozwala na całkowite wyleczenie[161]. Dla czerniaków o grubości nieprzekraczającej 1 mm zaleca się margines wycięcia o szerokości 1 cm, a szersze wycięcie jest zalecane w czerniakach o grubości 1,01–2 mm, które wymagają 2 cm marginesu zdrowych tkanek. Marginesy w trudnych lokalizacjach anatomicznych mogą być mniejsze od 2 cm, ale powinny być szersze niż 1 cm. W czerniaku in situ wystarcza margines 0,5–1 cm[162].
W kilku dużych badaniach oceniono skuteczność różnej szerokości zastosowanych marginesów chirurgicznych. Wykazano, że marginesy szersze niż 2 cm nie przynoszą dodatkowych korzyści dla chorych[163][164][165][166][167]. Metaanaliza wykazała również, że marginesy większe od 1 cm, ale mniejsze od 2 cm mogą być wystarczające[168]. Dawniej wykonywano znacznie szersze resekcje i standardem leczenia były marginesy o wielkości 3–5 cm, jednak późniejsze badania wykazały, że wznowy nie są związane ze zbyt małym marginesem wycięcia, a raczej z przerzutami drogą naczyń chłonnych[161]. W miarę możliwości linia cięcia powinna przebiegać w linii spływu chłonki[161].
W czerniaku wywodzącym się z plamy soczewicowatej może występować rozrost atypowych melanocytów poza widoczny obręb zmiany. Wykazano, że margines wycięcia 10 mm wystarcza do usunięcia 99% czerniaków z plamy soczewicowatej[169].
Czerniak podpaznokciowy wymaga przeprowadzenia dystalnej amputacji paliczka w stawie międzypaliczkowym lub amputacji całego palca[161].
Głębokość wycięcia powinna wynosić przynajmniej 1 cm, jednak w niektórych lokalizacjach musiałoby się wiązać to z wycięciem części mięśnia. Resekcja może być wykonana do naturalnej bariery jaką jest powięź[161].
Alternatywne metody do chirurgicznego wycięcia
Standardową metodą leczenia czerniaka in situ jest jego chirurgiczne wycięcie. U starannie wybranych chorych dopuszczalne jest miejscowe leczenie imikwimodem lub za pomocą radioterapii[162].
Biopsja węzła wartowniczego
Węzeł wartowniczy jest to pierwszy węzeł chłonny na drodze spływu chłonki z obszaru nowotworu, zwykle jest to pierwsza lokalizacja przerzutów nowotworowych drogą chłonną. Jednak brak obecności przerzutu w węźle wartowniczym nie pozwala wykluczyć obecności przerzutów węzłowych i odległych, które również mogą powstawać na drodze krwionośnej. Stwierdzanie zajęcia węzła wartowniczego ma znaczenie przy wyborze najkorzystniejszego leczenia[170].
Biopsja węzła wartowniczego jest wykonywana bezpośrednio po radykalnym wycięciu blizny po guzie pierwotnym po wykonanej wcześniej biopsji wycinającej czerniaka[4]. Wykorzystuje się dwie metody diagnostyczne: limfoscyntygrafię i metodę barwnikową. Limfoscyntygrafia polega na śródskórnym podaniu radioaktywnego znacznika znakowanego 99Tc we wszystkie cztery ćwiartki wyciętej blizny po czerniaku. W ciągu 1–30 minut wartownicze węzły chłonne są możliwe do zidentyfikowania za pomocą gammakamery, która wykrywa promieniowanie radioaktywnego znacznika zgromadzonego w węźle chłonnym. Po 4 godzinach radionuklid jest dystrybuowany do dalszych węzłów i tymczasowo nie ma możliwości rozpoznania węzła wartowniczego. Metoda barwnikowa polega na podaniu niebieskiego znacznika po obu stronach blizny. Wartowniczy węzeł chłonny gromadzi niebieski barwnik i jest wizualnie widoczny podczas operacji[171].
Biopsja węzła wartowniczego jest ważnym badaniem pozwalającym ocenić zaawansowanie choroby (stadiowanie), jednak wykazano niewielki jej wpływ na przeżycie chorych. Wieloletnia obserwacja zapoczątkowana już w 1994 roku wykazała, że biopsja węzła wartowniczego nie poprawia przeżycia specyficznego dla nowotworu (DSS) w całej populacji chorych na czerniaka. Jednak wykazano poprawę dziesięcioletniego przeżycia specyficznego dla nowotworu w grupie o pośredniej grubości guza (1,20–3,50 mm) z 41% do 56%. Poprawy przeżycia nie zaobserwowano u chorych z grubym czerniakiem (3,50 mm)[172]. Z kolei w przypadku cienkich czerniaków rzadko obserwuje się przerzuty węzłowe i rola biopsji węzła wartowniczego nie jest jasna. Prawdopodobieństwo zajęcia węzłów chłonnych, a tym samym zasadność wykonania zabiegu, zależą od czynników ryzyka obejmujących owrzodzenie i wysoki indeks mitotyczny. Ryzyko zajęcia węzłów chłonnych w czerniaku <0,75 mm wynosi 2,7%, a w czerniaku o grubości 0,75–1,00 mm ryzyko to wynosi około 6,2%[173]. W innym badaniu ryzyko zajęcia węzłów chłonnych w czerniaku poniżej 1,00 mm wynosiło poniżej 5%, ale w przypadku występowania owrzodzenia, wysokiego indeksu mitotycznego, regresji, IV stopnia według Clarka ryzyko to wynosiło 18%[174].
Nie zaleca się wykonywania biopsji węzła wartowniczego u chorych w stadium in situ, a także w stadium IA i IB zaawansowania klinicznego z bardzo cienkim czerniakiem poniżej 0,75 mm. Biopsja węzła wartowniczego może być rozważana u chorych z czerniakiem o grubości 0,75–1 mm[175]. U chorych w stadium IB i II zaawansowania klinicznego w guzach o grubości powyżej 1 mm lub w guzach o grubości 0,75–1 mm z owrzodzeniem lub indeksem mitotycznym ≥1/mm² zaleca się wykonanie biopsji węzła wartowniczego[175][176]. Biopsję należy rozważyć u chorych z resekowalnym przerzutem in transit w stadium III zaawansowania klinicznego, choć biopsja może być użyteczna w ocenie zaawansowania klinicznego, to jednak jej wpływ na przeżycie pozostaje niejasny[175].
Wycięcie węzłów chłonnych

Regionalna limfadenektomia jest zabiegiem usunięcia regionalnych węzłów chłonnych w celu poprawy lokalnej kontroli chorych i redukcji ryzyka wystąpienia przerzutów odległych, a tym samym poprawie przeżycia całkowitego. Zabieg polega na usunięciu możliwie wszystkich węzłów chłonnych w dorzeczu spływu chłonki obejmującym czerniaka. Wykazano, że zajęcie węzła wartowniczego jest związane z około 18% ryzykiem zajęcia kolejnych węzłów chłonnych[177][178][179].
Zwykle jest wykonywana w obrębie szyi, pachy i pachwiny. W obrębie szyi usuwa się 5 poziomów węzłów chłonnych, w obrębie pachy 3 poziomy, w pachwinie węzły chłonne powierzchowne i głębokie pachwinowo-udowe i biodrowo-zasłonowe[161].
W przypadku ujemnego węzła wartowniczego resekcja węzłów chłonnych nie jest konieczna. U chorych zakwalifikowanych do stadium III zaawansowania choroby z powodu zajęcia węzła wartowniczego zaleca się wykonanie limfadektomii. Chorzy z klinicznie zajętymi lokalnymi węzłami chłonnymi bez radiologicznych dowodów obecności przerzutów odległych są leczeni szeroką resekcją blizny po guzie i kompletną limfadektomią dorzecza spływu chłonki[180].
Leczenie adiuwantowe
Leczenie adiuwantowe (uzupełniające) jest to leczenie choroby nowotworowej uzupełniające zasadnicze leczenie, w przypadku czerniaka szerokie wycięcie chirurgiczne zmiany. Ma ono na celu zmniejszenie ryzyka wznowy miejscowej lub wystąpienia przerzutów odległych i docelowo wydłużenie przeżycia całkowitego chorych.
Chorzy z czerniakiem in situ i we wczesnych stadiach czerniaka są wyleczeni po samym szerokim wycięciu czerniaka z odpowiednim marginesem zdrowych tkanek i nie wymagają oni leczenia adiuwantowego. Jednak chorzy ze zmianami desmoplastycznymi, szczególnie z dużym neurotropizmem i suboptymalnymi marginesami wycięcia, są w grupie zwiększonego ryzyka nawrotu i lokalna radioterapia może poprawić miejscową kontrolę choroby[181]. Chorzy w stadiach IB i IIA nie wymagają leczenia adiuwantowego (poza ewentualnymi badaniami klinicznymi) i wystarczającym postępowaniem jest obserwacja. W stadium IIB i IIC z ujemnymi węzłami chłonnymi chorzy mogą być obserwowani lub poddani leczeniu wysokimi dawkami interferonu α. W stadium III zarówno w przypadku zajęcia węzła wartowniczego lub klinicznego zajęcia dalszych lokalnych węzłów chłonnych stosuje się interferon α w wysokich dawkach lub interferon pegylowany. Adiuwantowa radioterapia może być konieczna w przypadku klinicznie zajętych węzłów chłonnych i wysokim ryzykiem nawrotu po leczeniu chirurgicznym. W stadium III z przerzutami in transit lub w stadium IV leczenie adiuwantowe nie jest zalecane[182].
- Interferon

Interferony są to cytokiny wytwarzane przez makrofagi, monocyty, limfocyty B i T, komórki NK, komórki nabłonkowe i fibroblasty przede wszystkim w odpowiedzi na zakażenie wirusowe w celu modulacji działania układu immunologicznego. Rekombinowane interferony, obok leczenia niektórych zakażeń wirusowych, w tym wirusowego zapalenia wątroby typu B i C, znalazły zastosowanie w leczeniu niektórych chorób nowotworowych, w tym w leczeniu czerniaka, raka nerki i przewlekłej białaczki szpikowej. W leczeniu znajduje zastosowanie pegylowany interferon lub interferon w wysokich dawkach. Pegylowany interferon jest białkiem zmodyfikowanym poprzez przyłączenie do niego glikolu polietylenowego, co zmienia jego właściwości farmakologiczne i wydłuża czas optymalnego stężenia terapeutycznego[183].
Skuteczność interferonu w leczeniu adiuwantowym była oceniana w kilku badaniach klinicznych. W badaniu ECOG 1684 leczenie adiuwantowe chorych w stadium IIb i III za pomocą interferonu alfa-2b w wysokich dawkach porównano z samą obserwacją. W 7-letnim okresie obserwacji stwierdzono poprawę przeżycia wolnego od nawrotu choroby (RFS, relapse free survival) z 1 roku dla obserwacji do 1,7 roku dla leczonych adiuwantowo wysokimi dawkami interferonu oraz zaobserwowano wydłużenia przeżycia całkowitego (OS) z 2,8 lat w grupie obserwowanych do 3,8 lat w grupie leczonych adiuwantowo interferonem w wysokich dawkach[184]. Jednak w obserwacji 12,6-letniej już nie zaobserwowano wydłużenia przeżycia całkowitego, choć leczenie adiuwantowe interferonem wpływało na poprawę przeżycia wolnego od nawrotu choroby[185][180]. Metaanaliza 14 badań na 8122 chorych o wysokim ryzyku wznowy otrzymujących adiuwantowo interferon wskazuje na wydłużenie u nich przeżycia wolnego od choroby (DFS) i przeżycia całkowitego (OS)[186][187].
W badaniu EORTC na 18991 chorych w stadium III leczonych pegylowanym interferonem alfa porównano z samą obserwacją. Wykazano, że pegylowany interferon wydłuża czteroletnie przeżycie wolne od nawrotu (RFS) z 38,9% do 45,6% bez znaczącego wpływu pegylowanego interferonu na przeżycie całkowite[188][189].
Leczenie interferonem jest dość toksyczną terapią, choć badania wskazują na wydłużenie przeżycia wolnego od nawrotu, to wpływ na przeżycie całkowite nie jest jasny, dlatego w zaleceniach NCCN terapia otrzymała klasę zaleceń IIB[189]. Interferon alfa-2b w wysokich dawkach lub interferon pegylowany jest zalecany u chorych w stadium III zaawansowania klinicznego (zajęcie regionalnych węzłów chłonnych) oraz w niższych stadiach IIB i IIC (bez zajęcia węzłów chłonnych)[182]. Interferon w wysokich dawkach jest podawany przez 1 rok, a pegylowany przez 5 lat[189].
- Radioterapia
Radioterapia jest zalecana u chorych z czerniakiem desmoplastycznym z silnym neurotropizmem, szczególnie w warunkach suboptymalnego wycięcia zmiany, ponieważ zmniejsza odsetek nawrotów miejscowych[190][189].
Adiuwantowa radioterapia bywa stosowana u chorych z klinicznie zajętymi węzłami chłonnymi po całkowitym wycięciu zmiany z zajętymi węzłami w celu zmniejszenia ryzyka wznowy w węzłach chłonnych. Przed zastosowaniem metody rozważa się potencjalne korzyści związane z leczeniem oraz jej toksyczność i pogorszenie jakości życia[191].
W badaniu klinicznym Agrawal i współpracowników adiuwantowa radioterapia u chorych z zaawansowanym czerniakiem bez przerzutów odległych z wysokim ryzykiem nawrotów węzłowych zredukowała ona ryzyko nawrotu z 40,6% do 10,2% w pięcioletniej obserwacji. Jednocześnie zauważono znaczny wzrost chorobowości związanej z leczeniem[192]. W innym badaniu wykazano, że adiuwantowa radioterapia redukuje ryzyko nawrotu czerniaka w węzłach chłonnych[193], jednak jednocześnie dane wskazują na pogorszenie przeżycia u leczonych tą metodą[181].
Leczenie choroby zaawansowanej miejscowo, przerzutów in transit oraz wznowy
Guzki satelitarne (satelitoza), przerzuty in transit i wznowa stanowią pewne kontinuum szerzenia się nowotworu drogą limfatyczną, które jest związane ze złym rokowaniem[3][194].
Guzki satelitarne
Chorzy z guzkami satelitarnymi są leczeni według postępowania dla III stadium klinicznego choroby. Pierwotny guz jest usuwany za pomocą szerokiego wycięcia guza z ewentualną resekcją węzła wartowniczego lub pełną limfadenektomią[194]. Leczenie może być uzupełnione o INF-α lub radioterapię adiuwantową[191][195].
Przerzuty in transit

Istnieje kilka metod leczenia chorych z przerzutami in transit, jednak nie wypracowano optymalnej metody postępowania. Jeśli jest możliwe guz pierwotny i przerzut in transit jest wycinany w zakresie marginesu zdrowych tkanek. W niektórych przypadkach przeprowadza się biopsję węzła wartowniczego[196].
W leczeniu bywa stosowana terapia miejscowa za pomocą wstrzyknięć do guza szczepionki BCG, IL-2 lub INF-α. Zmiany bardzo powierzchowne niesięgające tkanki podskórnej mogą być leczone imikwimodem. Guz może być zniszczony za pomocą ablacji laserem. Radioterapia jest stosowana w nieoperacyjnym objawowym nawrocie[196].
Izolowana perfuzja i izolowana infuzja są technikami lokalnej dotętniczej podaży chemioterapii, która pozwala osiągać wysokie stężenie cytostatyków bez ogólnoustrojowych efektów toksyczności leczenia. Zwykle stosuje się melfalan. Izolowana infuzja kończyny jest techniką prostszą niż izolowana perfuzja, a pozwala osiągnąć podobny odsetek odpowiedzi[197][196]. W badaniu klinicznym na 128 chorych leczonych izolowaną infuzją osiągnięto 31% odsetek odpowiedzi całkowitych (CR)[198]. W modyfikacji tej metody z łagodną hipertermią osiągnięto 63% odsetek odpowiedzi całkowitych i 38% odsetek przeżycia pięcioletniego u leczonych[199].
Inną opcją terapeutyczną jest ogólnoustrojowe leczenie przerzutów in transit, szczególnie w przypadku postępu mimo leczenia regionalnego[200].
Wznowa w bliźnie
Wyróżnia się dwie odmienne sytuacje prowadzące do wznowy w bliźnie: zbyt oszczędne wycięcie guza pierwotnego oraz lokalny nawrót mimo dostatecznie szerokiego marginesu chirurgicznego zdrowych tkanek. Nawrót wymaga potwierdzenia histopatologicznego[201].
W przypadku zbyt wąskiego marginesu i wznowy leczenie polega na powtórnym wycięciu blizny z guzem. W przypadku nawrotu mimo odpowiedniego szerokiego marginesu tkanek zaleca się wycięcie guza z (lub bez) usunięcia regionalnych węzłów chłonnych[201].
Wznowa lokalna, satelitarna i in transit
Nawrót powinien być potwierdzony histopatologicznie na podstawie biopsji chirurgicznej lub biopsji cienkoigłowej. Wznowę resekuje się w miarę możliwości z marginesem zdrowych tkanek i wykonuje się biopsje węzła wartowniczego[201].
W nieoperacyjnym czerniaku in transit przeprowadza się izolowaną infuzje lub perfuzje kończyny lekiem cytostatycznym. Alternatywami są wstrzyknięcia szczepionki BCG, IL-2 lub INF-α, radioterapia, ablacja laserowa, a w powierzchownych zmianach bywa wykorzystywany imikwimod[202].
Po osiągnięciu całkowitej remisji chorych obserwuje się lub podaje adiuwantowo duże dawki interferonu α[202].
Wznowa węzłowa
Potwierdzenie nawrotu uzyskuje się za pomocą biopsji cienkoigłowej lub biopsji całego węzła chłonnego i badania histopatologicznego. U chorych z niecałkowitą resekcją węzłów chłonnych przeprowadza się powtórny zabieg w celu wycięcia całego dorzecza węzłowego drenującego chłonkę z okolicy guza. Po całkowitej resekcji przeprowadza się resekcję nawrotu węzłowego z odpowiednim marginesem zdrowych tkanek. Leczenie wznowy może być uzupełnione o terapię adiuwantową w postaci wysokich dawek interferonu α lub w wybranych przypadkach radioterapii. Jeśli całkowita resekcja jest niemożliwa, chory jest dalej leczony ogólnoustrojowo na zasadach leczenia choroby nieoperacyjnej i zaawansowanej[202].
Leczenie choroby zaawansowanej

.jpg)
Leczenie zaawansowanego czerniaka opiera się na leczeniu ogólnoustrojowym lub najlepszej terapii wspomagającej u chorych w złym stanie ogólnym. Istnieje wiele opcji leczenia ogólnoustrojowego, obejmujących nowe leki celowane, immunoterapię oraz klasyczną chemioterapię. Wykorzystanie niektórych leków jest zasadne wyłącznie przy odpowiednio wysokiej ekspresji cząstek, która warunkuje przewagę komórek nowotworowych nad prawidłowymi lub pomaga uniknąć odpowiedzi immunologicznej oraz kontrolowanej śmierci komórek. Wybór leczenia jest uzależniony od obecności mutacji BRAF, przebiegiem choroby nowotworowej i założonych celów leczenia, które może być nastawione na długotrwałą stabilizację choroby lub przede wszystkim na złagodzenie objawów[203]. Lekami pierwszego rzutu w ogólnoustrojowym leczeniu zaawansowanego czerniaka są wemurafenib lub dabrafenib (inhibitory BRAF), trametynib (inhibitor MEK), ipilimumab (przeciwciało anty CTLA-4), pembrolizumab i niwolumab (przeciwciała anty-PD-1/PD-L1) oraz IL-2 w wysokiej dawce[204].
W przypadku obecności mutacji BRAF zaleca się leczeniem wemurafenibem lub dabrafenibem (inhibitory BRAF). Inhibitory BRAF mogą być stosowane w połączeniu z trametynibem (inhibitor MEK) lub w monoterapii. Połączenie inhibitora BRAF z inhibitorem MEK skuteczniej wydłuża przeżycie leczonych niż monoterapia BRAF[203]. Trametynib nie jest zalecany w monoterapii poza nietolerancją inhibitorów BRAF[203].
Chorzy z niewielką objętością guza i bez objawów choroby są dobrymi kandydatami do immunoterapii w pierwszej linii leczenia za pomocą ipilimumabu (przeciwciało anty-CTLA-4) lub pembrolizumabu/niwolumabu (przeciwciała anty-PD-1/PD-L1) albo IL-2, która może wywołać trwałą odpowiedź i znacznie wydłużyć przeżycie leczonych[205][206]. Również w sytuacji braku mutacji BRAF stosuje się immunoterapię w leczeniu pierwszego rzutu. Jednak objawowa choroba lub progresja mino immunoterapii u chorych z mutacją BRAF skłania do zastosowania inhibitorów BRAF z lub bez inhibitora MEK[206].
Ipilimumab i pembrolizumab/niwolumab pozwalają osiągnąć niższy odsetek odpowiedzi obiektywnych niż inhibitory BRAF/BRAF+MEK. Dodatkowo później obserwuje się efekty kliniczne ipilimumabu niż inhibitorów BRAF/BRAF+MEK, ponieważ odbudowa odpowiedzi immunologicznej przeciw guzowi, która jest efektem działania ipilimumabu, wymaga czasu i regresja guza występuje później niż w przypadku inhibitorów BRAF/BRAF+MEK. Z drugiej strony ipilimumab i pembrolizumab/niwolumab wykazują większą zdolność do wywołania długoterminowej trwałej odpowiedzi przeciw nowotworowi i oferują większy odsetek długotrwałego przeżycia. Są one bardziej przydatne w leczeniu, które ma na celu długą stabilizację choroby i znaczne wydłużenie przeżycia[205]. Chorzy z przewidywalnym krótkim przeżyciem, z dużą masą guza, przerzutami w krytycznych lokalizacjach lub wywołujących uciążliwe objawy prawdopodobnie nie odniosą korzyści z leczenia przeciwciałami anty-CTLA-4 lub anty-PD-1/PD-L1 z powodu długiego czasu koniecznego do osiągnięcia wymiernych efektów klinicznych i w tym przypadku skuteczniejsze będzie leczenie inhibitorami BRAF, które szybciej i częściej wywołują odpowiedź na leczenie[205][207].
Pembrolizumab albo niwolumab mogą być stosowane w przypadku nieskuteczności ipilimumabu po leczeniu inhibitorem BRAF (jeśli występuje mutacja BRAF) w leczeniu drugiej linii lub w leczeniu pierwszej linii bez poprzedzającego leczenia ipilimumabem lub inhibitorem BRAF[208]. W przypadku stosunkowo rzadkiej obecności mutacji c-KIT podaje się imatynib[203]. Biochemioterapia lub klasyczna chemioterapia oparta na lekach alkilujących, przede wszystkim na dakarbazynie, jest jedną z opcji terapeutycznych stosowaną w leczeniu II linii, a u niektórych chorych w leczeniu I linii, szczególnie przy braku odpowiednich mutacji uzasadniających zastosowanie leków celowanych[209].
U części chorych wykonuje się operacje wycięcia korzystnie zlokalizowanych przerzutów (metastazektomia) lub operacje paliatywne w celu łagodzenia dolegliwości. Operuje się również w sytuacjach bezpośredniego zagrożenia życia w związku z chirurgicznymi powikłaniami choroby. Stosuje się paliatywną radioterapię objawowych przerzutów, szczególnie w obrębie mózgu[210].
Inhibitory BRAF
BRAF jest kinazą seroninową-treoninową aktywującą kinazy MAP/ERK. Gen jest zaliczany do protoonkogenów. Aktywność kinazy jest większa niż pozostałych kinaz z tej rodziny i jej mutacja aktywująca (onkogen) jest stosunkowo często obecna w licznych typach nowotworów[211]. Mutacja aktywująca genu BRAF jest związana z patogenezą czerniaka poprzez konstytutywną aktywację szlaku MEK/ERK, a w konsekwencji unikanie apoptozy, zwiększenie potencjału replikacyjnego, nasilenie angiogenezy, zdolności do nacieku tkanek i do tworzenia przerzutów, a także unikania odpowiedzi immunologicznej[211]. Mutacja BRAF prowadzi do niekontrolowanego podziału komórek nowotworowych[212].
Mutacje BRAF pojawiają się w skórze nieuszkodzonej przez słońce, ale mutacje tego genu są rzadkie w czerniaku błon śluzowych i w obrębie kończyn[211][213]. Prawie 50–60% chorych na uogólnionego czerniaka wykazuje mutacje genu BRAF, a najczęstszą z nich jest mutacja kodonu V600E (75% mutacji)[211][214], w której występuje zastąpienie waliny przez glutaminę. W mutacji V600K występuje lizyna w pozycji waliny[215]. Inhibitory BRAF powodują zmniejszenie masy guza, jednak odpowiedź na leczenie zwykle jest czasowa[216]. Do selektywnych inhibitorów BRAF należy wemurafenib oraz dabrafenib. Nieselektywnym inhibitorem BRAF jest sorafenib[215].
Inhibitory BRAF w połączeniu z inhibitorami MEK w porównaniu do monoterapii inhibitorem BRAF wykazują wyższy odsetek przeżycia całkowitego oraz wyższy odsetek odpowiedzi obiektywnych sięgający 50–75%[207][217][218]. Terapia inhibitorami BRAF oferuje stosunkowo wczesny początek regresji nowotworu, dlatego mogą być stosowane w przypadku szybko rozwijającej się choroby[207].
Niestety u większości chorych pojawia się oporność na leczenie inhibitorami BRAF i wówczas dochodzi do ponownego wzrostu guza. Mimo tego terapia u znacznej części leczonych pozwala na znaczne wydłużenie przeżycia[207]. Prawdopodobnie dalsze przedłużone stosowanie inhibitorów BRAF pomimo progresji wciąż pozwala przedłużyć przeżycie leczonych[219][220].
Wemurafenib jest wysoce specyficznym inhibitorem BRAF[221]. W badaniu klinicznym II fazy na 132 chorych na wcześniej leczonego uogólnionego czerniaka z mutacją BRAF lek wykazał 53% odsetek odpowiedzi obiektywnej (ORR), mediana czasu przeżycia wolnego od progresji (PFS) wynosiła 6,8 miesiąca, a mediana przeżycia całkowitego (OS) wynosiła 15,9 miesięcy[222]. Gorszą odpowiedź na leczenie zaobserwowano u chorych z podniesionym poziomem dehydrogenazy mleczanowej (LDH) 1,5-krotnie powyżej górnej granicy normy[223].
W badaniu klinicznym III fazy 2107 chorych losowo przydzielono do leczenia za pomocą wemurafenibu lub dakarbazyny. Sześciomiesięczny odsetek przeżycia całkowitego leczonych wemurafenibem wyniósł 84%, podczas gdy leczonych dakarbazyną 64%. Również mediana przeżycia całkowitego była lepsza u leczonych wemurafenibem niż dakarbazyną i wynosiła odpowiednio 13,2 miesiąca oraz 9,6 miesięcy. Odsetek odpowiedzi wynosił 48%[224].
- Dabrafenib
Dabrafenib jest inhibitorem BRAF. W badaniu klinicznym II fazy oceniono skuteczność leku na 92 chorych na zaawansowanego czerniaka z mutacjami BRAF V600E i mutacją V600K nieleczonych wcześniej lub leczonych inhibitorami MEK. W przypadku mutacji BRAF V600E odsetek odpowiedzi całkowitych (CR) wynosił 7% i 53% leczonych osiągnęło częściową odpowiedź (PR), ale był on słabszy u pacjentów z mutacją BRAF V600K, gdzie nie osiągnięto całkowitych odpowiedzi, a 13% osiągnęło częściową odpowiedź. Mediana czasu przeżycia wolnego od progresji choroby (PFS) była dłuższa dla dabrafenibu w porównaniu do standardowej chemioterapii. Dla mutacji BRAF V600E wynosiła ona 27 tygodni, a dla mutacji BRAF V600K 20 tygodni[225]. W badaniu klinicznym III fazy 250 chorych w stadium IV i nieoperacyjnym stadium III losowo przydzielono do terapii dabrafenibem lub dakarbazyną. Dabrafenib wykazał dłuższą medianę czasu przeżycia wolnego od progresji choroby, która dla dabrafenibu wynosiła 6,7 miesięcy, a dla dakarbazyny 2,9 miesięcy. Odsetek odpowiedzi obiektywnych w przypadku inhibitora BRAF wynosił 50%, a dla dakarbazyny tylko 6%[215].
W dużym polskim badaniu klinicznym z udziałem 704 chorych z przerzutowym czerniakiem porównano skuteczność połączenia dabrafenibu z trametynibem z monoterapią wenurafenibem. Wykazano, że połączenie dabrafenibu z trametynibem wywołuje lepszy odsetek 12-miesięcznego przeżycia całkowitego niż wenurafenib w monoterapii i wynosił on dla połączenia dabrafenibu z trametynibem 72% i 65% dla wenurafenibu w monoterapii. Również mediana przeżycia wolnego od progresji choroby była wyższa w połączeniu dabrafenibu z trametynibem (11,4 miesiąca) niż dla wenurafenibu (7,3 miesiąca)[217][226]. W badaniu III fazy połączenie dabrafenibu z tremetinibem wykazało wyższy niż dabrafenib w monoterapii odsetek 6-miesięcznego przeżycia całkowitego wynoszący 93%. Połączenie wykazało również wyższy odsetek obiektywnych odpowiedzi i medianę przeżycia wolnego od progresji choroby[218][226]. Połączenie dabrafenibu i trametynibu jest zalecane przy obecności mutacji BRAF, jeśli jest ono tolerowane przez chorego[226].
Inhibitory MEK
Białka MEK (MEK1 i MEK2) są elementami szlaku MAPK/ERK. Aktywowane BRAF fosforyluje białka MEK1 i MEK2, które aktywują kinazy MAP. Szlak MAP wpływa na zwiększenie przeżywalności komórek nowotworowych w wielu chorobach rozrostowych[227].
- Trametynib
Jest wysoce selektywnym przeciwciałem skierowanym przeciw białkom MEK[221]. Lek wywołuje słabszą poprawę przeżycia całkowitego i czasu wolnego od progresji choroby niż inhibitory BRAF (wenurafenib, dabrafenib), dlatego lek w monoterapii jest zarezerwowany dla chorych z mutacją BRAF i jednoczesną nietolerancją inhibitorów BRAF[226].
W badaniu klinicznym III fazy METRIC na 322 chorych porównano skuteczność trametynibu z klasyczną chemioterapią u chorych z mutacją BRAF V600E lub V600K. Odsetek obiektywnej odpowiedzi dla trametynibu wynosił 24% i dla chemioterapii 7%, z kolei mediana przeżycia wolnego od progresji choroby dla leku osiągnęła 4,8 miesięcy, a dla chemioterapii 1,4 miesiąca[227].
Połączenie trametynibu z dabrafenibem ma przewagę nad pojedynczym inhibitorem BRAF i może być stosowane u chorych tolerujących to połączenie[226]. Wykazano, że połączenie dabrafenibu z trametynibem zwiększa odsetek 6-miesięcznego przeżycia całkowitego w porównaniu z monoterapią wenurafenibem[217]. Trametynib dodany do dabrafenibu zwiększa odsetek 6-miesięcznego przeżycia całkowitego w porównaniu z samym dabrafenibem[218].
- Selumetynib
Jest to wysoce selektywny inhibitor MEK. W badaniu II fazy nie wykazano zwiększenia przeżycia całkowitego po dodaniu selumetynibu do dakarbazyny, choć czas przeżycia wolny od progresji był znacząco dłuższy przy leczeniu selumetynibem niż dakarbazyną[228].
Inhibitory c-KIT
Gen c-KIT koduje receptor kinazy tyrozynowej, która może aktywować wiele szlaków w tym RAS-RAF-MEK-ERK i PI3K-AKT[229]. Mutacja c-KIT nie jest częsta w czerniaku skórnym, w zmianach bez przewlekłego uszkodzenia słonecznego występuje jedynie z częstością 1–2%[229]. Jednak mutacja jest częstsza w czerniaku podpaznokciowo-kończynowym i czerniaku błon śluzowych, gdzie pojawia się w 20% tych nowotworów[230]. Mutacja ma istotne znaczenie w patogenezie tych typów czerniaka. Mutacja zwiększa potencjał proliferacyjny komórek nowotworowych i sprzyja migracji tych komórek, co z kolei sprzyja powstawaniu przerzutów[229].
- Imatynib
Imatynib jest inhibitorem kinazy tyrozynowej stosowanym w leczeniu przewlekłej białaczki szpikowej i guzów stomalnych (GIST). Lek jest skuteczny wyłącznie u chorych z mutacją c-KIT. W badaniu klinicznym na chorych z przerzutowym czerniakiem z mutacją c-KIT imatynib wykazał 23% odsetek odpowiedzi obiektywnych i 3,5 miesięczną medianę przeżycia wolnego od progresji choroby (PFS) i 51% osiągnęło 1 roczne przeżycie całkowite[231].
Immunoterapia
Czerniak wywołuje potencjalnie skuteczną przeciwnowotworową odpowiedź immunologiczną, jednak w mikrośrodowisku guza jest ona skutecznie hamowana za pomocą różnych niefizjologicznych mechanizmów generowanych przez komórki nowotworowe. W efekcie staje się ona niewydolna i nie pozwala na kontrolę wzrostu nowotworu. W celach terapeutycznych opracowano kilka metod neutralizacji mechanizmów blokady odpowiedzi immunologicznej[214]. W immunoterapii czerniaka stosuje się przeciwciała anty CTLA-4, przeciwciała anty-PD1 oraz IL-2 w wysokich dawkach[196].
Przeciwciała anty-PD1 i anty-PD-L1
Jest to grupa leków biologicznych będąca przeciwciałami skierowanymi przeciwko receptorowi programowanej śmierci PD-1 (ang. programmed cell death 1) oraz przeciwko jego ligandom PD-L1 i PD-L2. Fizjologicznie szlak PD1 jest odpowiedzialny za ograniczenie aktywności limfocytów T w celu redukcji nadmiernej odpowiedzi immunologicznej i reakcji autoimmunologicznych[232]. Jego ligandy PD-L1 i PD-L2 poprzez wpływ na kinazy wywołują zahamowanie szlaków pobudzających limfocyty T do aktywności i powodują ograniczenie aktywności tych komórek na różnych etapach odpowiedzi immunologicznej[233], głównie w etapie efektorowym[234].
W warunkach fizjologicznych mechanizmy pobudzające i hamujące odpowiedź immunologiczną pozostają we względnej równowadze, aby zapewnić odpowiednią reakcję na obce antygeny z jednoczesną tolerancją na antygeny własne (tolerancja immunologiczna). Jednak elementy mechanizmów hamujących odpowiedź mogą zostać patologicznie pozyskane przez komórki nowotworowe i przyczyniać się do unikania zniszczenia przez układ immunologiczny[234]. W wyniku nieprawidłowej regulacji w górę w komórkach czerniaka ligandy PD-L1 i PD-L2 mogą ulegać nadmiernej ekspresji[234][235], wówczas gdy ulegają one połączeniu z receptorem PD-1 limfocyt T otrzymuje silny sygnał hamujący blokujący jego aktywność przeciwnowotworową. Komórki czerniaka nabywają zdolność do wyłączenia odpowiedzi immunologicznej poprzez korzystną dla nich deregulację ekspresji ligandu PD-1, dopiero gdy pojawią się limfocyty T zdolne do skutecznej odpowiedzi przeciwnowotworowej[236]. Przewlekłe zahamowanie poprzez szlak PD-1 prowadzi do anergii limfocytów T i zahamowaniu wydajnej odpowiedzi immunologicznej przeciw guzowi[234].
- Pembrolizumab (MK-3475)
Pembrolizumab jest wysoce selektywnym przeciwciałem monoklonalnym skierowanym przeciw receptorowi PD-1. Jest wskazany u chorych, u których wystąpiła progresja choroby pomimo stosowania ipilimumabu i inhibitorów BRAF, jeśli stwierdzono jego mutację. W zaleceniach NCCN może być również stosowany jako leczenie pierwszego rzutu w chorobie uogólnionej lub nieresekowalnej[208].
Skuteczność leku oceniono w badaniu klinicznym KEYNOTE-001. W pierwszej fazie badania na 135 chorych wykazano 38% odsetek odpowiedzi obiektywnych, a mediana czasu przeżycia wolnego od progresji (PFS) wynosiła około 7 miesięcy. Nie było różnicy w odsetku odpowiedzi pomiędzy chorymi leczonymi wcześniej ipilimumabem a nieleczonymi nim[221][237][238]. W drugim etapie badania leku podano u 173 chorych z opornym na leczenie czerniakiem. Badanych podzielono na dwie grupy otrzymujące lek w różnych dawkach, odpowiednio 2 mg/kg i 10 mg/kg. W obu grupach osiągnięto ten sam odsetek odpowiedzi obiektywnych wynoszący 26%, mediana przeżycia wolnego od progresji choroby u chorych otrzymujących lek w dawce 2 mg/kg wynosiła 22 tygodnie, a u chorych otrzymujących lek w dawce 10 mg/kg 14 tygodni[221][238]. Ogólnie odsetek odpowiedzi u wszystkich leczonych w obu etapach badania wynosił 34%, choć w pewnych szczególnych grupach był on znacznie większy[238]. W badaniu KEYNOTE-002 na chorych wcześniej leczonych ipilimumabem oraz inhibitorem BRAF lub MEK porównano skuteczność pembrolizumabu z lekami cytostatycznymi. Odsetek odpowiedzi był wyższy niż w klasycznej chemioterapii i wynosił 21% u leczonych pembrolizumabem w dawce 2 mg/kg i 25% u leczonych dawką 10 mg/kg[238][239].
Niwolumab jest selektywnym przeciwciałem monoklonalnym przeciw receptorowi PD-1. Podobnie jak pembrolizumab jest wskazany u chorych, u których wystąpił progres choroby mimo stosowania ipilimumabu i inhibitorów BRAF, jeśli stwierdzono jego mutację. W zaleceniach NCCN może być stosowany jako leczenie pierwszego rzutu w chorobie uogólnionej lub nieresekowalnej[208].
W badaniu klinicznym I fazy wykazano, że lek wywołuje odpowiedź u 28% leczonych[240]. Odsetek przeżyć całkowitych po 1 roku wynosił 62%, a po 2 latach 43%. Mediana przeżyć całkowitych wynosiła 16,8 miesięcy, mediana czasu przeżycia wolnego od progresji wynosiła 3,7 miesięcy[241]. W badaniu klinicznym III fazy na 418 nieleczonych wcześniej chorych porównano skuteczność leku do dakarbazyny. Roczne przeżycie całkowite (OS) dla leczonych niwolumabem wynosiło 72,9%, a dla dakarbazyny 42,1%. Niwolumab w tym badaniu wykazał również lepszy profil bezpieczeństwa[221][242]. Testowano również połączenie niwolumabu z ipilimumabem. W badaniu na 80 chorych uzyskano 40% odsetek odpowiedzi obiektywnych i połączenie charakteryzuje się wyższym odsetkiem odpowiedzi obiektywnych niż te leki w monoterapii[243].
Przeciwciała anty-CTLA-4
CTLA-4 hamuje funkcję limfocytów T poprzez różne mechanizmy, w tym poprzez zapobieganie kostymulacji CD28 z jego ligandami CD80 i CD86, zablokowanie przekazywania sygnału z TCR/CD3[244] oraz zablokowanie cyklu komórkowego limfocytów, co uniemożliwia namnożenie się tych komórek[245].
W warunkach fizjologicznych aktywacja limfocytów T pociąga za sobą wzrost ekspresji CTLA-4 w ramach sprzężenia zwrotnego ujemnego i kontroli odpowiedzi immunologicznej[246]. W mikrośrodowisku guza CTLA-4 tłumi prawidłową aktywność przeciwnowotworową układu immunologicznego. Blokada CTLA-4 za pomocą przeciwciał monoklonalnych powoduje przywrócenie odpowiedzi przeciwnowotworowej. Sam mechanizm działania leków nie został w pełni wyjaśniony. Polega on nie tylko na zablokowaniu sygnału hamującego dla limfocytów T, ale również na zubożeniu populacji limfocytów T regulatorowych[247]. Zarejestrowanymi przeciwciałami przeciw CTLA-4 w leczeniu czerniaka są ipilimumab i tremelimumab[246].
Ipilimumab jest przeciwciałem monoklonalnym skierowanym przeciw CTLA-4 i jednym z leków ogólnoustrojowych pierwszego rzutu u chorych z zaawansowanym czerniakiem[203].
W badaniu klinicznym III fazy na 676 chorych porównano ipilimumab ze szczepionką przeciw gp100. Chorzy losowo otrzymali ipilimumab, szczepionkę przeciwnowotworową anty-gp100 lub połączenie szczepionki z iplimumabem. Mediana przeżycia całkowitego dla samego ipilimamubu wynosiła 10,1 miesiąca, dla połączenia ipilimumabu ze szczepionką anty-gp100 mediana przeżycia wynosiła 10,0 miesięcy, a dla samej szczepionki 6,4 miesięcy[248]. W innym badaniu porównano leczenie ipilimumabem w połączeniu z dakarbazyną i połączenia dakarbazyny z placebo. Połączenie ipilimumabu z dakarbazyną osiągnęło znacznie wyższą medianę przeżycia całkowitego wynoszącą 11,2 miesięcy niż dakarbazyna z placebo, dla której mediana przeżycia wynosiła 9,1 miesięcy[249]. W analizie danych z 12 badań klinicznych na łącznie 2985 chorych ipilimunab wykazał medianę przeżycia wynoszącą 11,4 miesiąca i 22% trzyletnie przeżycie całkowite[250].
- Tremelimumab
Tremelimumab jest przeciwciałem monoklonalnym przeciw CTLA-4. Badanie kliniczne III fazy porównujące skuteczność leku z klasyczną chemioterapią nie wykazało korzyści w odsetku odpowiedzi obiektywnej oraz odsetku przeżyć całkowitych w związku ze stosowaniem tremelimumabu[251].
Interleukina 2 (IL-2)
Interleukina 2 jest cytokiną stymulującą limfocyty T i komórki NK. W efekcie jej działania dochodzi do nasilenia odpowiedzi komórkowej[252]. Ze względu na znacznie nasiloną toksyczność terapii IL-2 może być stosowana jako alternatywa do innych terapii u chorych w dobrym stanie ogólnym[253]. Ryzyko powikłań wielonarządowych znacznie ogranicza zastosowanie leczenia do młodszej grupy chorych[252].
W badaniu klinicznym na 270 chorych wykazano, że IL-2 w wysokich dawkach wykazuje 16% odsetek odpowiedzi obiektywnych, 60% leczonych osiągnęło trwałą wielomiesięczną odpowiedź, odsetek całkowitych odpowiedzi wynosił 10%[254][255].
Szczepionka przeciwnowotworowa
Szczepionka przeciwnowotworowa jest lekiem podawanym w celach leczniczych (nie profilaktycznie przed zachorowaniem) mającym na celu ukierunkowanie odpowiedzi immunologicznej na rozpoznanie i zniszczenie komórek nowotworowych. Mimo licznych badań do tej pory nie uzyskano skutecznego preparatu zwiększającego przeżycie całkowite leczonych[256][257].
Badano liczne preparaty oparte o komórki i preparaty niezawierające komórek. Szczepionki zawierające komórki obejmowały preparaty zawierające lizaty komórek nowotworowych, całe komórki nowotworowe z adiuwantem immunologicznym, zmodyfikowane genetycznie całe komórki nowotworowe, komórki dendrytyczne stymulowane DNA, RNA, białkami lub lizaty komórek nowotworowych, a także limfocyty B. Preparaty niezawierające komórki były opierane o DNA komórek nowotworowych, białka komórek nowotworowych oraz wirusowych wektorach[256]. Nieskuteczność dotychczasowych szczepionek ma kilka przyczyn. Antygeny stosowane w szczepionkach są własnymi strukturami i limfocyty T zdolne do wysokiego powinowactwa do takich elementów są eliminowane w fizjologicznym mechanizmie tolerancji immunologicznej. Z kolei komórki nowotworowe są zdolne do umieszczenia na swojej powierzchni cząstek zdolnych do zahamowania odpowiedzi immunologicznej. Dodatkowo komórki nowotworowe wykazują klonalne zróżnicowanie, czyli nie stanowią jednorodnej grupy, wobec której można łatwo wypracować uniwersalne szczepienie przeciw konkretnej strukturze[257]. Obecne badania skupiają się na antygenach nowotworowych pojawiających się w każdej lub większości komórek nowotworowych bezpośrednio związanych ze wczesną patogenezą nowotworu[257].
W czerwcu 2021 do fazy II badań klinicznych trafił kandydat na szczepionkę mRNA firmy BioNTech, BNT111. Próby prowadzone był w 7 krajach, w tym w Polsce. Środek został podany, razem z cemiplimabem, 120 pacjentom z nieoperowalnym czerniakiem (reagującym na antyciało monoklonalne anti-PD-1, którym jest cemiplimab) w stadium III i IV.[258]
Kombinacje leków celowanych i immunomodulujących
- Połączenie inhibitorów BRAF i MEK
Połączenie inhibitora BRAF (dabrafenibu) i inhibirora MEK (trametynibu) jest jedną z zalecanych opcji leczniczych u chorych z zaawansowanym czerniakiem[204].
W jeszcze niezakończonym badaniu III fazy Combi-D leczenie chorych za pomocą kombinacji dabrafenibu i trametynibu porównano z monoterapią dabrafenibem. Wykazano, że kombinacja inhibitorów BRAF i MEK zwiększa odsetek przeżycia całkowitego w porównaniu z samodzielnym leczeniem inhibitorem BRAF[218]. Wstępne zaktualizowane wyniki badania wskazują, że mediana przeżycia leczonych połączeniem dabrafenibu i trametynibu wynosi 25,1 miesiąca, a 51% leczonych osiąga przeżycie dwuletnie[259][260].
Badane są również nowsze inhibitory MEK. W badaniu CoBRIM porównano połączenie wemurafenibu (inhibitor BRAF) i cobimetinibu (inhibitor MEK) z monoterapią wemurafenibem. Wstępne wyniki wskazują, że połączenie wemurafenibu i cobimetinibu wywołuje dłuższe przeżycie wolne od progresji choroby (PFS), którego mediana wynosiła 12,3 miesięcy niż monoterapia wemurafenibem z medianą przeżycia 7,2 miesięcy[260].
- Połączenie przeciwciał anty-CTLA-4 i anty-PD1/PD-L1
W trakcie badań oceniana jest przydatność połączenia przeciwciał anty-CTLA-4 i anty-PD1/PD-L1 w leczeniu przerzutowego czerniaka. Prawdopodobnie podwójna blokada wykazuje synergie. Blokada CTLA-4 prowadzi do aktywacji naiwnych limfocytów, a blokada PD-1 zapobiega ograniczeniu aktywności aktywnych limfocytów[205].
W randomizowanym badaniu klinicznym na 945 chorych z zaawansowanym czerniakiem porównano skuteczność połączenia ipilimumabu i niwolumabu z monoterapią ipilimumabem lub monoterapią niwolumabem. Badanie wskazuje, że połączenie ipilimumabu i niwolumabu w porównaniu do niwolumabu w monoterapii wywołuje dłuższe przeżycie wolne od progresji choroby. W przypadku nowotworów PD-L1-ujemnych blokada CTLA-4 i PD-L1 była bardziej skuteczna niż te leki w monoterapii. Ponadto odsetek odpowiedzi obiektywnych był wyższy w przypadku połączenia niż gdy leki były stosowane w monoterapii[261]. Jednak badanie ma pewne ograniczenia. Nieznana jest zdolność do wydłużenia przeżycia całkowitego (OS), a w grupie badanych występował znacznie niższy odsetek ekspresji PD-L1, co wyklucza z niego znaczną populację, która mogłaby skorzystać na monoterapii niwolumabem. Połączeniu towarzyszy większa toksyczność niż w przypadku monoterapii. Sugeruje to możliwość stosowania połączenia w przypadku nowotworów PD-L1 ujemnych, a monoterapii anty-PD1 w przypadku występowania PD-L1, ponieważ może ona wywoływać podobne przeżycie wolne od progresji choroby z mniejszymi efektami toksycznymi[260]. Zalecenia NCCN nie uwzględniają połączenia ipilimumabu z niwolumabem w leczeniu zaawansowanego czerniaka[204].
Chemioterapia

Czerniak jest nowotworem o stosunkowo dużej oporności na klasyczną chemioterapię. Mechanizmy oporności są podobne do innych nowotworów litych i wynikają z dużej bazowej oporności tkanki. W czerniaku występuje duża ekspresja enzymów naprawiających DNA i białek usuwających ksenobiotyki, co znacząco utrudnia działanie cytostatyków, dodatkowo aktywność szlaku kinazy aktywowana mitogenami i kinazy fosfatydyloinozytolu zwiększa oporność nowotworu na chemioterapię[262].
W chemioterapii zaawansowanego czerniaka wykorzystuje się dakarbazynę, temozolomid, paklitaksel, nab-paklitaksel i karboplatynę w monoterapii lub w połączeniach. W połączeniu z immunoterapią stosuje się dakarbazynę lub temozolomid z cisplatyną i dakarbazyną w połączeniu z IL-2 lub INF-α[263].
- Dakarbazyna
Dakarbazyna jest lekiem alkilującym DNA, cytostatyk jest standardem klasycznej chemioterapii w leczeniu zaawansowanego czerniaka, a przed wprowadzeniem nowoczesnych leków celowanych był lekiem pierwszego rzutu w tym stadium choroby. W największym badaniu klinicznym oceniającym ten lek wywołał on odpowiedź obiektywną u 7,5% leczonych[264]. Analiza 23 randomizowanych badań na łącznej liczbie 1390 leczonych dakarbazyną w monoterapii odsetek odpowiedzi obiektywnych wynosił 15,3%[265]. Odpowiedzi rzadko są trwałe[252]. Mediana przeżycia wolna od progresji choroby (PFS) w różnych badaniach waha się na poziomie 1,5-2 miesięcy, a mediana przeżycia całkowitego (OS) około 6-9 miesięcy[266][266][267][268]. Nigdy nie przeprowadzono randomizowanych badań porównujących skuteczność dakarbazyny do najlepszej terapii wspomagającej[268].
- Temozolomid
Temozolomid jest doustnym analogiem dakarbazyny ulegającym metabolizmowi do czynnego związku alkilującego MTIC[268]. Lek nie wykazuje przewagi nad dakarbazyną i wybór pomiędzy nimi jest związany z różną drogą podania, kosztami oraz obecnością przerzutów do mózgu[252].
W badaniu klinicznym porównano na 859 chorych temozolomid z dakarbazyną. Nie stwierdzono istotnych różnic w medianie przeżycia całkowitego wynoszącego dla dakarbazyny 9,4 miesiąca i dla temozolomidu 9,1 miesięcy. Również nie zauważono istotnych różnic w czasie przeżycia wolnego od progresji, wynoszącym odpowiednio 2,1 miesiąca i 2,3 miesiąca, a także odsetku odpowiedzi obiektywnych (odpowiednio 14% i 10%)[269]. W innym badaniu na 305 chorych również porównano temozolomid z dakarbazyną i nie wykazano statystycznie istotnych różnic pomiędzy odsetkiem odpowiedzi obiektywnych i przeżyciem całkowitym pomiędzy oboma lekami[252][266]. Lek dobrze penetruje do mózgu i jest przydatny w leczeniu przerzutów do ośrodkowego układu nerwowego, jednak w badaniu porównującym skuteczność połączenia temozolomidu z IL-2 i cisplatyną z połączeniem dakarbazyny z IL-2 i cisplatyną nie wykazano by temozolomid zapobiegał powstawaniu przerzutów w ośrodkowym układzie nerwowym[252].
- Alkaloidy Vinca i taksany
Alkaloidy Vinca (winblastyna, windezyna) i taksany (paklitaksel, docetaksel) w monoterapii wykazują skromną aktywność przeciwko czerniakowi. Winblastyna jest używana w połączeniu z innymi cytostatykami, przede wszystkim z cisplatyną i dakarbazyną w programie CVD z lub bez IL-2 lub INF-α[270][263] lub w połączeniu cisplatyny i temozolomidu z lub bez IL-2 lub INF-α[271][263]. Paklitaksel i nab-paklitaksel są stosowane w monoterapii lub w połączeniu z innymi lekami przeciwnowotworowymi[263]. Odsetek odpowiedzi obiektywnych paklitakselu w monoterapii lub w połączeniu z innymi lekami w różnych badaniach waha się pomiędzy 16 a 26%[272][273][274]. Paklitaksel jest stosowany w połączeniu z karboplatyną, ale schemat nie poprawia przeżycia leczonych, dlatego nie jest programem pierwszego rzutu[275][263].
- Kompleksy platyny
W leczeniu chorych na zaawansowanego czerniaka stosuje się połączenie paklitakselu i karboplatyny po nieskuteczności programów pierwszego rzutu[252][275]. W leczeniu karboplatyną obserwuje się 19% odsetek odpowiedzi obiektywnych[252][276], z kolei cisplatyna wywołuje 10% odsetek odpowiedzi obiektywnych[252][273].
- Pochodne nitrozomocznika
Pochodne nitrozomocznika (karmustyna, lomustyna, fotemustyna) wykazują podobną skuteczność jak dakarbazyna, ale wiążą się z większym stopniem toksyczności. Fotemustyna dość dobrze penetruje barierę krew-mózg i może być przydatna u chorych z przerzutami do mózgu[277]. W badaniu klinicznym na 219 chorych z przerzutowym czerniakiem porównano skuteczność fotemustyny z dakarbazyną. Wykazano, że leczeni fotemustyną osiągnęli lepszą medianę przeżycia całkowitego wynoszącą 7,3 miesięcy w porównaniu do dakarbazyny, gdzie mediana przeżycia całkowitego wynosiła 5,6 miesięcy[252][278].
- Biochemioterapia
Biochemioterapia jest połączeniem klasycznych leków przeciwnowotworowych z lekami biologicznymi. W leczeniu czerniaka znajduje zastosowanie połączenie programu CVD (cisplatyna, winblastyna, dakarbazyna) z IL-2 lub INF-α.
W małym badaniu klinicznym III fazy wykazano, że połączenie schematu CVD z IL-2 i INF-α wykazało większy odsetek odpowiedzi obiektywnych i medianę przeżycia całkowitego (OS) niż schemat CVD podawany samodzielnie[279]. W innym badaniu klinicznym połączenie CVD z IL-2 i INF-α dawało większy odsetek odpowiedzi obiektywnych i medianę czasu wolnego od progresji (PFS) niż CVD bez cytokin. Jednak połączenie z cytokinami nie wywoływało większego odsetka przeżyć całkowitych i jest leczeniem znacznie bardziej toksycznym niż CVD[280].
Metaanaliza na łącznie ponad 2600 leczonych chemioterapią z INF-α lub chemioterapią z INF-α i IL-2 potwierdziła, że biochemioterapia, choć podwyższa odsetek odpowiedzi obiektywnych i czas wolny od progresji choroby nie prowadzi do zwiększenia przeżycia całkowitego[281].
Leczenie chirurgiczne choroby zaawansowanej
- Metastazektomia
U starannie wybranych chorych w stadium IV zaawansowania klinicznego przy ograniczonej liczbie przerzutów przeprowadza się ich resekcję (metastazektomia)[200]. Metastazektomia nie jest wykonywana jeśli nie można usunąć wszystkich przerzutów. Choroba musi być ograniczona do pojedynczych obszarów o niewielkiej ilości przerzutów, a przerzuty muszą być korzystnie zlokalizowane. Najczęściej wycinane są łatwo dostępne przerzuty do skóry lub tkanki podskórnej, a rzadziej do niektórych narządów wewnętrznych[282]. Ograniczeniem metody jest częste występowanie ukrytych mikroprzerzutów towarzyszących wykrywalnym przerzutom[283]. Ważna jest nieobecność poważnych chorób współistniejących, które ograniczają korzyści z obciążającego leczenia chirurgicznego[284]. Leczenie nie może pogarszać jakości życia leczonych[282]. Paliatywna resekcja przerzutów w niektórych lokalizacjach u niektórych chorych może wydłużyć przeżycie nawet po nawrocie choroby nowotworowej[285][252][284][286].
- Paliatywne leczenie chirurgiczne
Paliatywne leczenie chirurgiczne nie jest przeprowadzane z intencją całkowitego wyleczenia czerniaka, ma ono na celu usunięcie lub złagodzenie uciążliwych objawów choroby nowotworowej, a więc poprawę jakości życia chorego. Paliatywne leczenie chirurgiczne znajduje zastosowanie w przypadku[285]:
- niedrożności przewodu pokarmowego,
- krwawieniu do przewodu pokarmowego powodującego niedokrwistość,
- objawowego krwawienia z przerzutów w innych lokalizacjach anatomicznych,
- przerzutów do skóry z owrzodzeniem lub dolegliwościami bólowymi,
- przerzutów do ośrodkowego układu nerwowego powodujących objawy neurologiczne,
- przerzutów do węzłów chłonnych lub innych lokalizacji powodujących objawy neurologiczne.
Radioterapia paliatywna
Czerniak jest uważany za nowotwór radiooporny. Radioterapia może przynosić korzyści chorym z objawowymi przerzutami, szczególnie w obrębie ośrodkowego układu nerwowego, gdzie bywa stosowana radioterapia stereotaktyczna (nóż gamma). Bywa stosowana również w innych lokalizacjach w celu łagodzenia uciążliwych objawów zaawansowanej choroby nowotworowej[283]. Napromieniowywanie zwykle jest połączone z innym leczeniem ogólnoustrojowym[252].
Rokowanie
| Stadium kliniczne | Przeżycie całkowite | |||
| guz bez owrzodzenia | guz z owrzodzeniem | |||
| IA | T1a | 95% | – | – |
| IB | T2a | 89% | T1b | 91% |
| IIA | T3a | 79% | T2b | 77% |
| IIB | T4a | 67% | T3b | 63% |
| IIC | – | – | T4b | 45% |
| IIIA | N1a N2a | 67% | – | – |
| IIIB | N1b N2b | 54% | N1a N2a | 52% |
| IIIC | N3 | 28% | N1b N2b N3 | 24% |
| Stadium kliniczne | Przeżycie całkowite |
| M1a | 62% |
| M1b | 53% |
| M1c | 32% |
Czerniak pozaskórny
Czerniak w większości przypadków jest zlokalizowany w obrębie skóry, która stanowi ponad 90–95% lokalizacji tego nowotworu. Pewna część guzów powstaje w rzadszych lokalizacjach pozaskórnych. Około 3,7% guzów lokalizuje się w obrębie narządu wzroku, 1,4% w obrębie błon śluzowych[289][290][291].
Czerniak narządu wzroku


Czerniak oka jest rzadkim nowotworem, jednak stanowi drugą po skórze lokalizację czerniaka. Łącznie stanowi 3,7% przypadków czerniaka u ludzi[292]. W 95% przypadków czerniak pojawia się w obrębie błony naczyniowej obejmującą naczyniówkę, ciało rzęskowe i tęczówkę[293]. W 90% przypadków czerniak oka dotyczy naczyniówki, w 3–4% ciała rzęskowego i tęczówki[294]. Spojówka jest lokalizacją czerniaka narządu wzroku tylko w 5% przypadków tego nowotworu[295]. Czynniki ryzyka są mało poznane, a rola promieniowania UV w powstawaniu czerniaka oka jest niejednoznaczna[293][292].
Objawy choroby są zróżnicowane w zależności od lokalizacji guza. Czerniak naczyniówki objawia się zaburzeniami widzenia, ubytkami w polu widzenia, błyskami w polu widzenia (fotopsje), krzywieniem się obrazu (metamorfopsje), bólem oka i mętami. W miarę postępu choroby dochodzi do odwarstwienia siatkówki, jaskry neowaskularnej i zapalenia błony naczyniowej oka, które ostatecznie prowadzą do jednoocznej ślepoty chorego oka[292].
Czerniak ciała rzęskowego często cechuje się długim bezobjawowym przebiegiem, następnie pojawiają się zaburzenia akomodacji, refrakcji, zlokalizowana zaćma lub zwiększone ciśnienie wewnątrz gałki ocznej spowodowane zamknięciem kąta przesączania[292].
Czerniak spojówki objawia się obecnością uniesionej, pogrubiałej i zabarwionej zmiany z gęstą siecią naczyń i melanozą. Zabarwienie może być w różnym odcieniu, a w 20% czerniaka spojówki jest on bezbarwny (czerniak amelanocytarny)[295].
Podstawą rozpoznania jest stwierdzenie obecności guza w badaniu okulistycznym za pomocą lampy szczelinowej i ewentualnie trójlustra Goldmana w pewnych lokalizacjach. Pomocniczo stosuje się ultrasonografię, angiografię fluoresceinową, koherencyjną tomografię optyczną i biomikroskopię ultradźwiękową[296]. Ostateczne rozpoznanie jest stawiane na podstawie oceny histopatologicznej materiału po wycięciu zmiany[297].
W etapie choroby ograniczonej wyłącznie do oka leczenie polega na brachyterapii, chirurgicznej resekcji guza lub enukleacji oka, przezźreniczej termoterapii albo terapii fotodynamicznej[298].
Enukleacja jest operacją usunięcia gałki ocznej z pozostawieniem zawartości oczodołu (w przeciwieństwie do wypatroszenia oczodołu). Jest metodą wykorzystywaną przede wszystkim w stadiach zaawansowanych choroby, kiedy nowotwór powoduje nieodwracalną ślepotę chorego oka. W korzystnych lokalizacjach guza przeprowadza się resekcję zmiany zwykle wspomaganych uzupełniającą radioterapią[298].
Alternatywą do leczenia chirurgicznego jest brachyterapia, polegająca na precyzyjnym umieszczeniu za pomocą aplikatora w okolicy guza radioaktywnych izotopów106Ru emitującego promieniowanie beta lub 125I emitującego głównie promieniowanie gamma[298]. Badanie COMS wykazało brak przewagi okaleczającej operacji nad brachyterapią[299]. Brachyterapię cechuje niski wskaźnik nawrotów wynoszący 3% po 7 latach[300]. Jednak wyniki terapii determinuje precyzyjne umieszczenie płytki z izotopem w okolicy guza[298].
Przezźrenicza termoterapia jest metodą wykorzystującą laser emitujący promieniowanie podczerwone, który wywołuje martwicę napromieniowywanego guza. Samodzielnie stosowana przezźrenicza termoterapia cechuje się znacznie większym odsetkiem nawrotów niż brachyterapia[301][302][303], jednak prawdopodobnie połączenie obu metod może dawać korzyści[304].
Leczenie choroby uogólnionej jest głównie oparte o chemioterapię. Wykorzystuje się dakarbazyne, temozolomid z lub bez INF-α, fotemustyne, karboplatyne w połączeniu z akarbamazyną z IFN-alfa i IL-2[298]. Jednak obserwowany odsetek odpowiedzi obiektywnych jest niewielki[298].
Mutacja c-KIT jest częsta w czerniaku narządu wzroku i może dotyczyć 50% guzów pierwotnych i 75% nowotworów przerzutowych[305]. Jednak badania kliniczne na imatynibie nie potwierdzają skuteczności tego leku[305][306]. Mutacje BRAF są rzadkie w czerniaku oka. Badania wskazują na pewną skuteczność trametynibu w leczeniu czerniaka naczyniówki[307][308]. Podobnie nie potwierdzono skuteczności ipilimumabu, przeciwciała anty-CTLA-4, w leczeniu czerniaka oka[309].
Czerniak błon śluzowych


Czerniak błon śluzowych jest rzadką postacią czerniaka i stanowi około 1,4% wszystkich przypadków czerniaka[290][291]. Średni wiek zachorowania jest o dekadę wyższy niż w przypadku czerniaka skóry, większość chorych skończyło 65. rok życia. Ze względu na znaczny udział czerniaka błon śluzowych w lokalizacji żeńskiego układu moczowo-płciowego częściej chorują kobiety, jednak poza tym narządem rozpowszechnianie choroby jest podobne u obu płci[290]. Czynniki ryzyka zachorowanie są niepoznane i prawdopodobnie promieniowanie UV nie odgrywa tak znaczącej roli jak w lokalizacji w obrębie skóry[290].
Czerniak błon śluzowych jest zlokalizowany w obrębie błon śluzowych przewodu pokarmowego, głowy i szyi oraz układu moczowo-płciowego. Przede wszystkim dotyczy odbytnicy i kanału odbytu, jamy ustnej, sromu i pochwy. Objawy są uzależnione od lokalizacji guza[290].
Rozpoznanie ze względu na skąpoobjawowy i niecharakterystyczny przebieg jest późne, choroba zwykle jest rozpoznawana w bardzo zaawansowanym stadium. Wstępne rozpoznanie jest stawiane na podstawie badania lekarskiego, badań obrazowych i endoskopowych. Ostateczne rozpoznanie jest stawiane na podstawie badania histopatologicznego materiału pobranego drogą biopsji. Kluczowe jest odróżnienie czerniaka błon śluzowych od znacznie powszechniejszych przerzutów do błon śluzowych. Znacznym utrudnieniem diagnostycznym bywa obecność przerzutów przy jednoczesnej regresji guza pierwotnego, który wówczas pozostaje nieuchwytny[290].
Leczenie czerniaka błon śluzowych jest oparte o chirurgiczną resekcję guza z szerokimi marginesami zdrowych tkanek[291][310]. Zwykle po zabiegu przeprowadza się adiuwantową radioterapię. W leczeniu adiuwantowym stosuje się interferon α. Nie ma badań oceniających skuteczność leczenia adiuwantowego. W chorobie uogólnionej stosuje się terapię celowaną, immunoterapię i klasyczną chemioterapię.
Wykazano, że w czerniaku błon śluzowych również może dochodzić do wykorzystania CTLA-4 do tłumienia odpowiedzi immunologicznej[311]. Aktywność w czerniaku błon śluzowych wykazuje przeciwciało anty-CTLA4 – ipilimumab. W badaniu na 71 chorych z przerzutowym czerniakiem błon śluzowych leczonych ipilimumabem zaobserwowano 12% odsetek odpowiedzi, a mediana czasu wolnego od progresji choroby (PFS) wynosiła 4,3 miesięcy i mediana przeżycia całkowitego 6,4 miesięcy[312].
W 20–40% przypadków czerniaka błon śluzowych występuje mutacja genu kinany tyrozynowej c-KIT[230][313]. Skutecznym lekiem w leczeniu aktywującej mutacji c-KIT jest Imatynib. Badania przedkliniczne wykazały skuteczność imatynibu[314][315][316]. W badaniu II fazy na 43 chorych na czerniaka, z czego 11 chorowało na czerniaka błony śluzowej, leczeni imatynibem osiągnęli 3,5 miesięczną medianę czasu wolnego od progresji choroby[231].
Do tej pory nie opublikowano badań oceniających skuteczność inhibitorów BRAF (wemurafenib, dabrafenib), inhibitorów MEK (trametynib, selumetynib) i przeciwciał przeciwko PD-1 (pembrolizumab i niwolumab) w leczeniu przerzutowego czerniaka błon śluzowych[317]. Kilka małych badań wskazuje na potencjalną przydatność ewerolimusu (inhibitor kinazy mTOR)[317][318][319].
W chemioterapii choroby zaawansowanej stosuje się leki stosowane w leczeniu czerniaka skóry. Wykorzystuje się dakarbazynę, temozolomid, połączenie dakarbazyny, cisplatyna, karmustyny i tamoksyfenu, a także biochemioterapię z IL-2 i INF-α[291][317].
Czerniak u zwierząt
Czerniak został stwierdzony u niektórych gatunków zwierząt dzikich, domowych i o znaczeniu gospodarczym. Częstość występowania czerniaka jest różna pośród różnych gatunków zwierząt. Dotyczy przede wszystkim koni, bydła, psów i świń[320][321][322]. Z kolei nowotwór jest rzadki u kotów, kóz i ptaków[320][323]. Prawdopodobnie w patogenezie czerniaka u zwierząt promieniowanie ultrafioletowe pełni mniejszą rolę[324].
Rozpoznanie jest stawiane na podstawie obrazu klinicznego i badania histopatologicznego. Pomocne może być wykonanie biopsji. Leczenie polega na usunięciu zmiany z odpowiednio szerokim marginesem zdrowych tkanek. W leczeniu bywa stosowana radioterapia i chemioterapia[325][326][327].
Historia

Pierwszy opis czerniaka pojawił się w pracy Hipokratesa, kolejny opis podał Rufus z Efezu[328]. Pierwsze współczesne opisy choroby podali Highmore (1651), Bonet (1651), Henrici i Nothnagel (1757), którzy zaobserwowali czarny nowotwór dający czarne przerzuty. W 1787 roku John Hunter pierwszy usunął guz, który później rozpoznano jako czerniak. W 1804 roku Rene Laennec jako pierwszy wyodrębnił czerniaka jako osobną chorobę. W 1820 roku William Norris opublikował pierwszy szczegółowy opis choroby, podając opis przebiegu choroby wywodzącej się ze znamienia i wynik sekcji zwłok. Zwrócił on uwagę na intensywny rozsiew przerzutów do licznych lokalizacji. Norris opisał również postać amelanocytarną czerniaka. W 1838 Robert Carswell wyróżnił podstawowe postacie choroby i określił ją słowem melanoma. W 1966 roku Wallace Clark opracował rokowniczą klasyfikację czerniaków w oparciu o badanie histopatologiczne. W 1970 roku Alexander Breslow stwierdził, że rokowanie czerniaka jest najbardziej uzależnione od wielkości i stopnia inwazji guza[328].
Pod koniec XIX wieku leczenie opierało się o wycięcie za pomocą skalpela lub nożyczek albo przyżeganie guza pierwotnego za pomocą środków żrących. Opracowanie skuteczniejszych metod stało się możliwe po wprowadzeniu znieczulenia ogólnego. W 1845 roku po raz pierwszy przeprowadzono operację całkowitego usunięcia guza pierwotnego. W 1892 Herbert Snow przeprowadził operację wycięcia czerniaka usuwając również węzły chłonne. W 1905 roku William Anderson Handley zaproponował zabieg polegający na usunięciu guza z marginesem tkanek o grubości 5 cm, z wycięciem tkanki podskórnej i powięzi, a także węzłów chłonnych[328].
Chemioterapię wprowadzono do leczenia nowotworów na początku lat pięćdziesiątych. W 1968 roku opublikowano wyniki badań wskazujące, że lek alkilujący melfalan wykazuje aktywność w przerzutowym czerniaku, jednak jego skuteczność była krótkotrwała i terapia wykazywała znaczną toksyczność[329]. W 1972 roku wprowadzono do leczenia czerniaka dakarbazynę, która stała się standardem leczenia zaawansowanego czerniaka, jednak lek nie wykazuje wysokiej skuteczności w leczeniu choroby[330]. W latach 90. wprowadzono immunoterapię za pomocą IL-2 w wysokich dawkach[328]. Przełomem w leczeniu czerniaka stało się zarejestrowanie nowych leków celowanych: ipilimumabu i wemurafenibu w 2011 roku, dabrafenibu i trametynibu w 2013 roku, niwolumabu i pembrolizumabu w 2014 roku[221].
Uwagi
- ↑ Jest to wyrażenie pleonastyczne, gdyż nie istnieje czerniak niezłośliwy (łagodny).
Przypisy
- ↑ a b Szczeklik i Gajewski 2014 ↓, s. 2220.
- ↑ a b c d A.G. Goodson, D. Grossman. Strategies for early melanoma detection: Approaches to the patient with nevi. „Journal of the American Academy of Dermatology”. 60 (5), s. 719-735; quiz 736-8, May 2009. DOI: 10.1016/j.jaad.2008.10.065. PMID: 19389517.
- ↑ a b c d Włodzimierz Ruka, Maciej Krzakowski, Waldemar Placek, Piotr Rutkowski i inni. Czerniaki skóry – zasady postępowania diagnostyczno-terapeutycznego. „Onkologia w Praktyce Klinicznej”, 2009.
- ↑ a b c Piotr Rutkowski, Piotr J. Wysocki, Czerniaki skóry [dostęp 2015-09-13] [zarchiwizowane z adresu 2016-03-17].
- ↑ a b H. Tsao, C. Bevona, W. Goggins, T. Quinn. The transformation rate of moles (melanocytic nevi) into cutaneous melanoma: a population-based estimate. „Archives of dermatology”. 139 (3), s. 282–288, Mar 2003. PMID: 12622618.
- ↑ a b Mayo Clinic: Melanoma. [dostęp 2015-08-29]. [zarchiwizowane z tego adresu (2015-08-28)].
- ↑ Casciato i Territo 2012 ↓, s. 437.
- ↑ Morris 2003 ↓, s. 103.
- ↑ U. Kellner, N. Bornfeld, M.H. Foerster. Severe course of cutaneous melanoma associated paraneoplastic retinopathy. „British Journal of Ophthalmology”. 79 (8), s. 746–752, Aug 1995. PMID: 7547786.
- ↑ a b c d e f g A.I. Riker, N. Zea, T. Trinh. The epidemiology, prevention, and detection of melanoma. „The Ochsner Journal”. 10 (2), s. 56–65, 2010. PMID: 21603359.
- ↑ Mirosław Strączyński, Co to jest czerniak? | lek. med. Mirosław Strączyński, miroslawstraczynski.pl, 7 stycznia 2020 [dostęp 2020-01-10] [zarchiwizowane z adresu 2020-08-10] (pol.).
- ↑ a b c d e f g h i j k D.S. Rigel. Epidemiology of melanoma. „Semin Cutan Med Surg”. 29 (4), s. 204–209, Dec 2010. DOI: 10.1016/j.sder.2010.10.005. PMID: 21277533.
- ↑ a b c d e Dummer i in. 2011 ↓, s. 15.
- ↑ a b c d e M. Berwick, E. Erdei, J. Hay. Melanoma epidemiology and public health. „Dermatol Clin”. 27 (2), s. 205-214, viii, Apr 2009. DOI: 10.1016/j.det.2008.12.002. PMID: 19254665.
- ↑ a b c R.M. MacKie, A. Hauschild, A.M. Eggermont. Epidemiology of invasive cutaneous melanoma. „Ann Oncol”. 20 Suppl 6, s. vi1-7, Aug 2009. DOI: 10.1093/annonc/mdp252. PMID: 19617292.
- ↑ C.A. Fink, M.N. Bates. Melanoma and ionizing radiation: is there a causal relationship?. „Journal of Radiation Research”. 164 (5), s. 701–710, Nov 2005. PMID: 16238450.
- ↑ a b c Weedon i in. 2005 ↓, s. 54.
- ↑ F.R. de Gruijl, H.J. Sterenborg, P.D. Forbes, R.E. Davies i inni. Wavelength dependence of skin cancer induction by ultraviolet irradiation of albino hairless mice. „Cancer Research”. 53 (1), s. 53–60, Jan 1993. PMID: 8416751.
- ↑ T.R. Fears, C.C. Bird, D. Guerry, R.W. Sagebiel i inni. Average midrange ultraviolet radiation flux and time outdoors predict melanoma risk. „Cancer Research”. 62 (14), s. 3992–3996, Jul 2002. PMID: 12124332.
- ↑ J.M. Elwood, R.P. Gallagher. Body site distribution of cutaneous malignant melanoma in relationship to patterns of sun exposure. „International Journal of Cancer”. 78 (3), s. 276–280, Oct 1998. DOI: <276::AID-IJC2>3.0.CO;2-S 10.1002/(SICI)1097-0215(19981029)78:3<276::AID-IJC2>3.0.CO;2-S. PMID: 9766557.
- ↑ a b P. Autier, J.F. Doré. Influence of sun exposures during childhood and during adulthood on melanoma risk. EPIMEL and EORTC Melanoma Cooperative Group. European Organisation for Research and Treatment of Cancer. „International Journal of Cancer”. 77 (4), s. 533–537, Aug 1998. PMID: 9679754.
- ↑ K.E. Johnson, B.C. Wulff, T.M. Oberyszyn, T.A. Wilgus. Ultraviolet light exposure stimulates HMGB1 release by keratinocytes. „Archives of Dermatological Research”. 305 (9), s. 805–815, Nov 2013. DOI: 10.1007/s00403-013-1401-2. PMID: 23942756.
- ↑ a b c Dummer i in. 2011 ↓, s. 16.
- ↑ A.J. Dobson, S.R. Leeder. Mortality from malignant melanoma in Australia: effects due to country of birth. „International Journal of Epidemiology”. 11 (3), s. 207–211, Sep 1982. PMID: 7129734.
- ↑ S. Gandini, F. Sera, M.S. Cattaruzza, P. Pasquini i inni. Meta-analysis of risk factors for cutaneous melanoma: II. Sun exposure. „European Journal of Cancer”. 41 (1), s. 45–60, Jan 2005. DOI: 10.1016/j.ejca.2004.10.016. PMID: 15617990.
- ↑ R.P. Gallagher, J.J. Spinelli, T.K. Lee. Tanning beds, sunlamps, and risk of cutaneous malignant melanoma. „Cancer Epidemiol Biomarkers Prev”. 14 (3), s. 562–566, Mar 2005. DOI: 10.1158/1055-9965.EPI-04-0564. PMID: 15767329.
- ↑ a b c Dummer i in. 2011 ↓, s. 17.
- ↑ a b c d J.H. Silva, B.C. Sá, A.L. Avila, G. Landman i inni. Atypical mole syndrome and dysplastic nevi: identification of populations at risk for developing melanoma - review article. „Clinics (Sao Paulo)”. 66 (3), s. 493–499, 2011. PMID: 21552679.
- ↑ a b c d e f Małgorzata Michalska-Jakubus, Tomasz Jakubus, Dorota Krasowska. Czerniak – epidemiologia, etiopatogeneza i rokowanie. „Medycyna Rodzinna”, 2006.
- ↑ T.M. Skender-Kalnenas, D.R. English, P.J. Heenan. Benign melanocytic lesions: risk markers or precursors of cutaneous melanoma?. „J Am Acad Dermatol”. 33 (6), s. 1000–1007, Dec 1995. PMID: 7490345.
- ↑ D. Massi, P. Carli, A. Franchi, M. Santucci. Naevus-associated melanomas: cause or chance?. „Melanoma Res”. 9 (1), s. 85–91, Feb 1999. PMID: 10338338.
- ↑ a b c Elder i Yun 2014 ↓, s. 32.
- ↑ a b c Massi i LeBoit 2013 ↓, s. 274.
- ↑ a b W.H. Clark, D.E. Elder, D. Guerry, M.N. Epstein i inni. A study of tumor progression: the precursor lesions of superficial spreading and nodular melanoma. „Hum Pathol”. 15 (12), s. 1147–1165, Dec 1984. PMID: 6500548.
- ↑ a b Weedon i in. 2005 ↓, s. 109.
- ↑ Mooi i Krausz 2007 ↓, s. 195.
- ↑ Daniel C. Chung, Daniel A. Haber: Principles of Clinical Cancer Genetics: A Handbook from the Massachusetts General Hospital. Springer Science & Business Media, 2010. ISBN 978-0-387-93846-2.
- ↑ Mooi i Krausz 2007 ↓, s. 198.
- ↑ a b A.K. Rustgi. Familial pancreatic cancer: genetic advances. „Genes & Development”. 28 (1), s. 1–7, Jan 2014. DOI: 10.1101/gad.228452.113. PMID: 24395243.
- ↑ H.T. Lynch, R.E. Brand, D. Hogg, C.A. Deters i inni. Phenotypic variation in eight extended CDKN2A germline mutation familial atypical multiple mole melanoma-pancreatic carcinoma-prone families: the familial atypical mole melanoma-pancreatic carcinoma syndrome. „Cancer”. 94 (1), s. 84–96, Jan 2002. PMID: 11815963.
- ↑ a b Weedon i in. 2005 ↓, s. 63.
- ↑ Dummer i in. 2011 ↓, s. 17–18.
- ↑ C.B. Begg, I. Orlow, A.J. Hummer, B.K. Armstrong i inni. Lifetime risk of melanoma in CDKN2A mutation carriers in a population-based sample. „J Natl Cancer Inst”. 97 (20), s. 1507–1515, Oct 2005. DOI: 10.1093/jnci/dji312. PMID: 16234564.
- ↑ a b Dummer i in. 2011 ↓, s. 18.
- ↑ S. Raimondi, F. Sera, S. Gandini, S. Iodice i inni. MC1R variants, melanoma and red hair color phenotype: a meta-analysis. „Int J Cancer”. 122 (12), s. 2753–2760, Jun 2008. DOI: 10.1002/ijc.23396. PMID: 18366057.
- ↑ D. Zhang, C. Chen, X. Fu, S. Gu i inni. A meta-analysis of DNA repair gene XPC polymorphisms and cancer risk. „J Hum Genet”. 53 (1), s. 18–33, 2008. DOI: 10.1007/s10038-007-0215-5. PMID: 18097734.
- ↑ S. Blankenburg, I.R. König, R. Moessner, P. Laspe i inni. Assessment of 3 xeroderma pigmentosum group C gene polymorphisms and risk of cutaneous melanoma: a case-control study. „Carcinogenesis”. 26 (6), s. 1085–1090, Jun 2005. DOI: 10.1093/carcin/bgi055. PMID: 15731165.
- ↑ a b R.C. Millikan, A. Hummer, C. Begg, J. Player i inni. Polymorphisms in nucleotide excision repair genes and risk of multiple primary melanoma: the Genes Environment and Melanoma Study. „Carcinogenesis”. 27 (3), s. 610–618, Mar 2006. DOI: 10.1093/carcin/bgi252. PMID: 16258177.
- ↑ J. Han, G.A. Colditz, J.S. Liu, D.J. Hunter. Genetic variation in XPD, sun exposure, and risk of skin cancer. „Cancer Epidemiol Biomarkers Prev”. 14 (6), s. 1539–1544, Jun 2005. DOI: 10.1158/1055-9965.EPI-04-0846. PMID: 15941969.
- ↑ T. Debniak, R.J. Scott, B. Górski, C. Cybulski i inni. Common variants of DNA repair genes and malignant melanoma. „Eur J Cancer”. 44 (1), s. 110–114, Jan 2008. DOI: 10.1016/j.ejca.2007.10.006. PMID: 18024013.
- ↑ C. Li, Z. Liu, L.E. Wang, J.E. Gershenwald i inni. Haplotype and genotypes of the VDR gene and cutaneous melanoma risk in non-Hispanic whites in Texas: a case-control study. „Int J Cancer”. 122 (9), s. 2077–2084, May 2008. DOI: 10.1002/ijc.23357. PMID: 18183598.
- ↑ a b c d Weedon i in. 2005 ↓, s. 55.
- ↑ F.J. Moloney, H. Comber, P. O’Lorcain, P. O’Kelly i inni. A population-based study of skin cancer incidence and prevalence in renal transplant recipients. „Br J Dermatol”. 154 (3), s. 498–504, Mar 2006. DOI: 10.1111/j.1365-2133.2005.07021.x. PMID: 16445782.
- ↑ a b c d e G. Chaidemenos, A. Stratigos, M. Papakonstantinou, F. Tsatsou. Prevention of malignant melanoma. „Hippokratia”. 12 (1), s. 17–21, Jan 2008. PMID: 18923759.
- ↑ H. Vainio, A.B. Miller, F. Bianchini. An international evaluation of the cancer-preventive potential of sunscreens. „Int J Cancer”. 88 (5), s. 838–842, Dec 2000. PMID: 11072258.
- ↑ M.P. Purdue, L. From, B.K. Armstrong, A. Kricker i inni. Etiologic and other factors predicting nevus-associated cutaneous malignant melanoma. „Cancer Epidemiol Biomarkers Prev”. 14 (8), s. 2015–2022, Aug 2005. DOI: 10.1158/1055-9965.EPI-05-0097. PMID: 16103454.
- ↑ L.K. Dennis, L.E. Beane Freeman, M.J. VanBeek. Sunscreen use and the risk for melanoma: a quantitative review. „Ann Intern Med”. 139 (12), s. 966–978, Dec 2003. PMID: 14678916.
- ↑ B.L. Diffey. Sunscreens and melanoma: the future looks bright. „Br J Dermatol”. 153 (2), s. 378–381, Aug 2005. DOI: 10.1111/j.1365-2133.2005.06729.x. PMID: 16086753.
- ↑ V.R. Doherty, R.M. MacKie. Reasons for poor prognosis in British patients with cutaneous malignant melanoma. „Br Med J (Clin Res Ed)”. 292 (6526), s. 987–989, Apr 1986. PMID: 3083979.
- ↑ N. Calonge, D.B. Petitti, T.G. DeWitt, L. Gordis i inni. Screening for skin cancer: U.S. Preventive Services Task Force recommendation statement. „Ann Intern Med”. 150 (3), s. 188–193, Feb 2009. PMID: 19189908.
- ↑ J.F. Aitken, P.H. Youl, M. Janda, J.B. Lowe i inni. Increase in skin cancer screening during a community-based randomized intervention trial. „Int J Cancer”. 118 (4), s. 1010–1016, Feb 2006. DOI: 10.1002/ijc.21455. PMID: 16152577.
- ↑ J.F. Aitken, J.M. Elwood, J.B. Lowe, D.W. Firman i inni. A randomised trial of population screening for melanoma. „J Med Screen”. 9 (1), s. 33–37, 2002. PMID: 11943795.
- ↑ a b B. Krone, K.F. Kölmel, J.M. Grange, G. Mastrangelo i inni. Impact of vaccinations and infectious diseases on the risk of melanoma - evaluation of an EORTC case-control study. „Eur J Cancer”. 39 (16), s. 2372–2378, Nov 2003. PMID: 14556930.
- ↑ A. Pfahlberg, K.F. Kölmel, J.M. Grange, G. Mastrangelo i inni. Inverse association between melanoma and previous vaccinations against tuberculosis and smallpox: results of the FEBIM study. „J Invest Dermatol”. 119 (3), s. 570–575, Sep 2002. DOI: 10.1046/j.1523-1747.2002.00643.x. PMID: 12230497.
- ↑ K.F. Kölmel, J.M. Grange, B. Krone, G. Mastrangelo i inni. Prior immunisation of patients with malignant melanoma with vaccinia or BCG is associated with better survival. An European Organization for Research and Treatment of Cancer cohort study on 542 patients. „Eur J Cancer”. 41 (1), s. 118–125, Jan 2005. DOI: 10.1016/j.ejca.2004.09.023. PMID: 15617996.
- ↑ B. Krone, K.F. Kölmel, B.M. Henz, J.M. Grange. Protection against melanoma by vaccination with Bacille Calmette-Guerin (BCG) and/or vaccinia: an epidemiology-based hypothesis on the nature of a melanoma risk factor and its immunological control. „Eur J Cancer”. 41 (1), s. 104–117, Jan 2005. DOI: 10.1016/j.ejca.2004.08.010. PMID: 15617995.
- ↑ a b Szczeklik i Gajewski 2014 ↓, s. 2218.
- ↑ a b Weedon i in. 2005 ↓, s. 52.
- ↑ Skin cancers. [dostęp 2015-09-13]. [zarchiwizowane z tego adresu (2015-06-03)].
- ↑ Weedon i in. 2005 ↓, s. 53.
- ↑ Dummer i in. 2011 ↓, s. 14.
- ↑ D.M. Parkin, F. Bray, J. Ferlay, P. Pisani. Estimating the world cancer burden: Globocan 2000. „Int J Cancer”. 94 (2), s. 153–156, Oct 2001. PMID: 11668491.
- ↑ R. Böni, C. Schuster, B. Nehrhoff, G. Burg. Epidemiology of skin cancer. „Neuro Endocrinol Lett”. 23 Suppl 2, s. 48–51, Jul 2002. PMID: 12163848.
- ↑ a b c Witold Kycler, Marek Teresiak. Czerniak skóry: aktualne możliwości leczenia w Polsce na podstawie analizy leczonych pacjentów i przeglądu literatury. „Współczesna Onkologia”, 2006. [dostęp 2017-10-24].
- ↑ Dummer i in. 2011 ↓, s. 13.
- ↑ Witold Zatoński, Joanna Didkowska, Urszula Wojciechowska, Nowotwory złośliwe w Polsce w 2011 roku [dostęp 2014-06-11].
- ↑ Dummer i in. 2011 ↓, s. 14–15.
- ↑ E. Erdei, S.M. Torres. A new understanding in the epidemiology of melanoma. „Expert Rev Anticancer Ther”. 10 (11), s. 1811–1823, Nov 2010. DOI: 10.1586/era.10.170. PMID: 21080806.
- ↑ Weedon i in. 2005 ↓, s. 50.
- ↑ a b H.S. Greenwald, E.B. Friedman, I. Osman. Superficial spreading and nodular melanoma are distinct biological entities: a challenge to the linear progression model. „Melanoma Res”. 22 (1), s. 1–8, Feb 2012. DOI: 10.1097/CMR.0b013e32834e6aa0. PMID: 22108608.
- ↑ a b c d e f g h i j k l m Weedon i in. 2005 ↓, s. 66.
- ↑ a b c d B.G. Goldstein, A.O. Goldstein. Diagnosis and management of malignant melanoma. „Am Fam Physician”. 63 (7), s. 1359–1368, 1374, Apr 2001. PMID: 11310650.
- ↑ a b Massi i LeBoit 2013 ↓, s. 445.
- ↑ Elder i Yun 2014 ↓, s. 3.
- ↑ a b Kumar, Cotran i Robins 2005 ↓, s. 916.
- ↑ a b c d e f Massi i LeBoit 2013 ↓, s. 446.
- ↑ a b c d e f Weedon i in. 2005 ↓, s. 68.
- ↑ a b c d e Massi i LeBoit 2013 ↓, s. 451.
- ↑ Weedon i in. 2005 ↓, s. 69.
- ↑ a b Mooi i Krausz 2007 ↓, s. 289.
- ↑ a b Massi i LeBoit 2013 ↓, s. 452.
- ↑ Stefan Kruś, Ewa Skrzypek-Fakhoury: Patomorfologia kliniczna. Warszawa: PZWL, 2007, s. 876. ISBN 978-83-200-3283-3.
- ↑ Weedon i in. 2005 ↓, s. 70.
- ↑ a b Mooi i Krausz 2007 ↓, s. 293.
- ↑ Weedon i in. 2005 ↓, s. 70–71.
- ↑ a b Weedon i in. 2005 ↓, s. 71.
- ↑ a b Weedon i in. 2005 ↓, s. 73.
- ↑ a b c d Weedon i in. 2005 ↓, s. 74.
- ↑ Weedon i in. 2005 ↓, s. 75.
- ↑ a b Weedon i in. 2005 ↓, s. 77.
- ↑ Weedon i in. 2005 ↓, s. 77–78.
- ↑ a b Weedon i in. 2005 ↓, s. 79.
- ↑ Stefania Jabłońska, Sławomir Majewski: Dermatologia. PZWL, 2008, s. 376. ISBN 978-83-200-3723-4.
- ↑ a b Weedon i in. 2005 ↓, s. 80.
- ↑ a b Weedon i in. 2005 ↓, s. 83.
- ↑ Weedon i in. 2005 ↓, s. 83–84.
- ↑ Weedon i in. 2005 ↓, s. 86.
- ↑ Weedon i in. 2005 ↓, s. 86–87.
- ↑ a b c d G. Palmieri, M. Capone, M.L. Ascierto, G. Gentilcore i inni. Main roads to melanoma. „Journal of Translational Medicine”. 7, s. 86, 2009. DOI: 10.1186/1479-5876-7-86. PMID: 19828018.
- ↑ Hanna Koseła, Tomasz Świtaj, Piotr Rutkowski. Zastosowanie inhibitorów BRAF i MEK w terapii zaawansowanego czerniaka. „Onkologia w Praktyce Klinicznej”, 2011.
- ↑ E. Shtivelman, M.Q. Davies, P. Hwu, J. Yang i inni. Pathways and therapeutic targets in melanoma. „Oncotarget”. 5 (7), s. 1701–1752, Apr 2014. PMID: 24743024.
- ↑ a b C. Bertolotto. Melanoma: from melanocyte to genetic alterations and clinical options. „Scientifica (Cairo)”. 2013, s. 635203, 2013. DOI: 10.1155/2013/635203. PMID: 24416617.
- ↑ M. Takata, H. Murata, T. Saida. Molecular pathogenesis of malignant melanoma: a different perspective from the studies of melanocytic nevus and acral melanoma. „Pigment Cell & Melanoma Research”. 23 (1), s. 64–71, Feb 2010. DOI: 10.1111/j.1755-148X.2009.00645.x. PMID: 19788535.
- ↑ H. Tsao, L. Chin, L.A. Garraway, D.E. Fisher. Melanoma: from mutations to medicine. „Genes & Development”. 26 (11), s. 1131–1155, Jun 2012. DOI: 10.1101/gad.191999.112. PMID: 22661227.
- ↑ J.M. Mehnert, H.M. Kluger. Driver mutations in melanoma: lessons learned from bench-to-bedside studies. „Curr Oncol Rep”. 14 (5), s. 449–457, Oct 2012. DOI: 10.1007/s11912-012-0249-5. PMID: 22723080.
- ↑ David DPolsky, Carlos Cordon-Cardo. Oncogenes in melanoma. „Oncogene”, 2003.
- ↑ a b c d e Weedon i in. 2005 ↓, s. 61.
- ↑ a b c L. Mervic. Time course and pattern of metastasis of cutaneous melanoma differ between men and women. „PLoS One”. 7 (3), s. e32955, 2012. DOI: 10.1371/journal.pone.0032955. PMID: 22412958.
- ↑ a b c F. Tas. Metastatic behavior in melanoma: timing, pattern, survival, and influencing factors. „Journal of Oncology”. 2012, s. 647684, 2012. DOI: 10.1155/2012/647684. PMID: 22792102.
- ↑ DeVita, Lawrence i Rosenberg 2008 ↓, s. 1912.
- ↑ F. Meier, S. Will, U. Ellwanger, B. Schlagenhauff i inni. Metastatic pathways and time courses in the orderly progression of cutaneous melanoma. „Br J Dermatol”. 147 (1), s. 62–70, Jul 2002. PMID: 12100186.
- ↑ DeVita, Lawrence i Rosenberg 2008 ↓, s. 1899.
- ↑ a b c d e f g h i j k Grażyna Kamińska-Winciorek, Radosław Śpiewak. Podstawy dermoskopii zmian melanocytowych dla początkujących. „Postępy Higieny i Medycyny Doświadczalnej”, 2011.
- ↑ a b c d A. Kardynal, M. Olszewska. Modern non-invasive diagnostic techniques in the detection of early cutaneous melanoma. „J Dermatol Case Rep”. 8 (1), s. 1–8, Mar 2014. DOI: 10.3315/jdcr.2014.1161. PMID: 24748903.
- ↑ a b c John i Stolz 2010 ↓, s. 7.
- ↑ a b A. Blum, R. Hofmann-Wellenhof, H. Luedtke, U. Ellwanger i inni. Value of the clinical history for different users of dermoscopy compared with results of digital image analysis. „Journal of the European Academy of Dermatology”. 18 (6), s. 665–669, Nov 2004. DOI: 10.1111/j.1468-3083.2004.01044.x. PMID: 15482291.
- ↑ John i Stolz 2010 ↓, s. 3.
- ↑ I. Tromme, L. Sacré, F. Hammouch, C. Legrand i inni. Availability of digital dermoscopy in daily practice dramatically reduces the number of excised melanocytic lesions: results from an observational study. „British Journal of Dermatology”. 167 (4), s. 778–786, Oct 2012. DOI: 10.1111/j.1365-2133.2012.11042.x. PMID: 22564185.
- ↑ a b c d John i Stolz 2010 ↓, s. 4.
- ↑ C. Longo, F. Farnetani, S. Ciardo, A.M. Cesinaro i inni. Is confocal microscopy a valuable tool in diagnosing nodular lesions? A study of 140 cases. „British Journal of Dermatology”. 169 (1), s. 58–67, Jul 2013. DOI: 10.1111/bjd.12259. PMID: 23374159.
- ↑ a b c d E. Szymańska, M. Maj, M. Majsterek, J. Litniewski i inni. The usefulness of high frequency ultrasonography in dermatological practice-ultrasound features of selected cutaneous lesions. „Polski Merkuriusz Lekarski”. 31 (181), s. 37–40, Jul 2011. PMID: 21870707.
- ↑ D. Jasaitiene, S. Valiukeviciene, G. Linkeviciute, R. Raisutis i inni. Principles of high-frequency ultrasonography for investigation of skin pathology. „Journal of European Academy of Dermatology and Venereology”. 25 (4), s. 375–382, Apr 2011. DOI: 10.1111/j.1468-3083.2010.03837.x. PMID: 20849441.
- ↑ a b c d DeVita, Lawrence i Rosenberg 2008 ↓, s. 1904.
- ↑ a b c d Coit i in. 2015 ↓, s. 40.
- ↑ Anna Nasierowska-Guttmejer, Barbara Górnicka: Zalecenia do diagnostyki histopatologicznej nowotworów. Warszawa: Centrum Onkologii, Oddział Gliwice; Polskie Towarzystwo Patologów, 2013, s. 19–21. ISBN 978-83-909137-1-1.
- ↑ a b c d e f P. Mohr, A.M. Eggermont, A. Hauschild, A. Buzaid. Staging of cutaneous melanoma. „Annals of Oncology”. 20 Suppl 6, s. vi14-21, Aug 2009. DOI: 10.1093/annonc/mdp256. PMID: 19617293.
- ↑ a b W.D. Holder, R.L. White, J.H. Zuger, E.J. Easton i inni. Effectiveness of positron emission tomography for the detection of melanoma metastases. „Ann Surg”. 227 (5), s. 764-769; discussion 769-71, May 1998. PMID: 9605668.
- ↑ D. Rinne, R.P. Baum, G. Hör, R. Kaufmann. Primary staging and follow-up of high risk melanoma patients with whole-body 18F-fluorodeoxyglucose positron emission tomography: results of a prospective study of 100 patients. „Cancer”. 82 (9), s. 1664–1671, May 1998. PMID: 9576286.
- ↑ H.C. Steinert, R.A. Huch Böni, A. Buck, R. Böni i inni. Malignant melanoma: staging with whole-body positron emission tomography and 2-[F-18]-fluoro-2-deoxy-D-glucose. „Radiology”. 195 (3), s. 705–709, Jun 1995. DOI: 10.1148/radiology.195.3.7753998. PMID: 7753998.
- ↑ S.M. Swetter, L.A. Carroll, D.L. Johnson, G.M. Segall. Positron emission tomography is superior to computed tomography for metastatic detection in melanoma patients. „Annals of Surgery Oncol”. 9 (7), s. 646–653, Aug 2002. PMID: 12167578.
- ↑ H.E. Daldrup-Link, C. Franzius, T.M. Link, D. Laukamp i inni. Whole-body MR imaging for detection of bone metastases in children and young adults: comparison with skeletal scintigraphy and FDG PET. „American Journal of Roentgenology”. 177 (1), s. 229–236, Jul 2001. DOI: 10.2214/ajr.177.1.1770229. PMID: 11418435.
- ↑ A. Aydin, J.Q. Yu, H. Zhuang, A. Alavi. Detection of bone marrow metastases by FDG-PET and missed by bone scintigraphy in widespread melanoma. „Clin Nucl Med”. 30 (9), s. 606–607, Sep 2005. PMID: 16100478.
- ↑ Cancer Research UK: Stages of melanoma. [dostęp 2015-07-10]. [zarchiwizowane z tego adresu (2015-07-10)].
- ↑ a b L. Schuchter, D.J. Schultz, M. Synnestvedt, B.J. Trock i inni. A prognostic model for predicting 10-year survival in patients with primary melanoma. The Pigmented Lesion Group. „Ann Intern Med”. 125 (5), s. 369–375, Sep 1996. PMID: 8702087.
- ↑ a b C.M. Balch, S.J. Soong, J.E. Gershenwald, J.F. Thompson i inni. Prognostic factors analysis of 17,600 melanoma patients: validation of the American Joint Committee on Cancer melanoma staging system. „Journal of Clinical Oncology”. 19 (16), s. 3622–3634, Aug 2001. PMID: 11504744.
- ↑ a b c d e f g h J. Homsi, M. Kashani-Sabet, J.L. Messina, A. Daud. Cutaneous melanoma: prognostic factors. „Cancer Control”. 12 (4), s. 223–229, Oct 2005. PMID: 16258493.
- ↑ a b Weedon i in. 2005 ↓, s. 64.
- ↑ C. Garbe, P. Büttner, J. Bertz, G. Burg i inni. Primary cutaneous melanoma. Identification of prognostic groups and estimation of individual prognosis for 5093 patients. „Cancer”. 75 (10), s. 2484–2491, May 1995. PMID: 7736392.
- ↑ A. Breslow. Thickness, cross-sectional areas and depth of invasion in the prognosis of cutaneous melanoma. „Annals of Surgery”. 172 (5), s. 902–908, Nov 1970. PMID: 5477666.
- ↑ Nicholas Reed, Green John Alan, David M. Gershenson, Nadeem Siddiqui, Rachel Connor: Rare and Uncommon Gynecological Cancers: A Clinical Guide. Springer Science & Business Media, 2010, s. 204. ISBN 978-3-642-13492-0.
- ↑ E. Nagore, V. Oliver, R. Botella-Estrada, S. Moreno-Picot i inni. Prognostic factors in localized invasive cutaneous melanoma: high value of mitotic rate, vascular invasion and microscopic satellitosis. „Melanoma Res”. 15 (3), s. 169–177, Jun 2005. PMID: 15917698.
- ↑ a b S.B. Kesmodel, G.C. Karakousis, J.D. Botbyl, R.J. Canter i inni. Mitotic rate as a predictor of sentinel lymph node positivity in patients with thin melanomas. „Annals of Surgical Oncology”. 12 (6), s. 449–458, Jun 2005. DOI: 10.1245/ASO.2005.04.027. PMID: 15864482.
- ↑ S.C. Paek, K.A. Griffith, T.M. Johnson, V.K. Sondak i inni. The impact of factors beyond Breslow depth on predicting sentinel lymph node positivity in melanoma. „Cancer”. 109 (1), s. 100–108, Jan 2007. DOI: 10.1002/cncr.22382. PMID: 17146784.
- ↑ V.K. Sondak, J.M. Taylor, M.S. Sabel, Y. Wang i inni. Mitotic rate and younger age are predictors of sentinel lymph node positivity: lessons learned from the generation of a probabilistic model. „Ann Surg Oncol”. 11 (3), s. 247–258, Mar 2004. PMID: 14993019.
- ↑ W.H. Clark, D.E. Elder, D. Guerry, L.E. Braitman i inni. Model predicting survival in stage I melanoma based on tumor progression. „J Natl Cancer Inst”. 81 (24), s. 1893–1904, Dec 1989. PMID: 2593166.
- ↑ V.J. McGovern, H.M. Shaw, G.W. Milton. Prognosis in patients with thin malignant melanoma: influence of regression. „Histopathology”. 7 (5), s. 673–680, Sep 1983. PMID: 6629343.
- ↑ a b c d Coit i in. 2015 ↓, s. 37.
- ↑ a b c d e f Czerniak skóry. Klasyfikacja TNM. [dostęp 2015-07-14]. [zarchiwizowane z tego adresu (2015-07-15)].
- ↑ Coit i in. 2015 ↓, s. 36.
- ↑ Dummer i in. 2012 ↓, s. 1.
- ↑ a b c d e f Sławomir Trepka, Piotr Rutkowski, Zbigniew I. Nowecki, Janusz Słuszniak. Chirurgia w leczeniu czerniaków. „Nowotwory”. 66, 2010.
- ↑ a b Coit i in. 2015 ↓, s. 46.
- ↑ U. Veronesi, N. Cascinelli. Narrow excision (1-cm margin). A safe procedure for thin cutaneous melanoma. „Arch Surg”. 126 (4), s. 438–441, Apr 1991. PMID: 2009058.
- ↑ U. Veronesi, N. Cascinelli, J. Adamus, C. Balch i inni. Thin stage I primary cutaneous malignant melanoma. Comparison of excision with margins of 1 or 3 cm. „N Engl J Med”. 318 (18), s. 1159–1162, May 1988. DOI: 10.1056/NEJM198805053181804. PMID: 3079582.
- ↑ C.M. Balch, S.J. Soong, T. Smith, M.I. Ross i inni. Long-term results of a prospective surgical trial comparing 2 cm vs. 4 cm excision margins for 740 patients with 1–4 mm melanomas. „Ann Surg Oncol”. 8 (2), s. 101–108, Mar 2001. PMID: 11258773.
- ↑ C.M. Balch, M.M. Urist, C.P. Karakousis, T.J. Smith i inni. Efficacy of 2-cm surgical margins for intermediate-thickness melanomas (1 to 4 mm). Results of a multi-institutional randomized surgical trial. „Ann Surg”. 218 (3), s. 262-267; discussion 267-9, Sep 1993. PMID: 8373269.
- ↑ J.M. Thomas, J. Newton-Bishop, R. A’Hern, G. Coombes i inni. Excision margins in high-risk malignant melanoma. „N Engl J Med”. 350 (8), s. 757–766, Feb 2004. DOI: 10.1056/NEJMoa030681. PMID: 14973217.
- ↑ P.I. Haigh, L.A. DiFronzo, D.R. McCready. Optimal excision margins for primary cutaneous melanoma: a systematic review and meta-analysis. „Can J Surg”. 46 (6), s. 419–426, Dec 2003. PMID: 14680348.
- ↑ J.H. Kunishige, D.G. Brodland, J.A. Zitelli. Surgical margins for melanoma in situ. „J Am Acad Dermatol”. 66 (3), s. 438–444, Mar 2012. DOI: 10.1016/j.jaad.2011.06.019. PMID: 22196979.
- ↑ Radzisław Kordek: Onkologia. Podręcznik dla studentów i lekarzy. Gdańsk: VIA MEDICA, 2007, s. 91. ISBN 978-83-7555-016-0.
- ↑ F. Amersi, D.L. Morton. The role of sentinel lymph node biopsy in the management of melanoma. „Adv Surg”. 41, s. 241–256, 2007. PMID: 17972569.
- ↑ D.L. Morton, J.F. Thompson, A.J. Cochran, N. Mozzillo i inni. Final trial report of sentinel-node biopsy versus nodal observation in melanoma. „N Engl J Med”. 370 (7), s. 599–609, Feb 2014. DOI: 10.1056/NEJMoa1310460. PMID: 24521106.
- ↑ R.H. Andtbacka, J.E. Gershenwald. Role of sentinel lymph node biopsy in patients with thin melanoma. „J Natl Compr Canc Netw”. 7 (3), s. 308–317, Mar 2009. PMID: 19401063.
- ↑ C. Mitteldorf, H.P. Bertsch, K. Jung, K.M. Thoms i inni. Sentinel node biopsy improves prognostic stratification in patients with thin (pT1) melanomas and an additional risk factor. „Ann Surg Oncol”. 21 (7), s. 2252–2258, Jul 2014. DOI: 10.1245/s10434-014-3641-6. PMID: 24652352.
- ↑ a b c Coit i in. 2015 ↓, s. 47–48.
- ↑ S.L. Wong, C.M. Balch, P. Hurley, S.S. Agarwala i inni. Sentinel lymph node biopsy for melanoma: American Society of Clinical Oncology and Society of Surgical Oncology joint clinical practice guideline. „J Clin Oncol”. 30 (23), s. 2912–2918, Aug 2012. DOI: 10.1200/JCO.2011.40.3519. PMID: 22778321.
- ↑ N. Cascinelli, E. Bombardieri, R. Bufalino, T. Camerini i inni. Sentinel and nonsentinel node status in stage IB and II melanoma patients: two-step prognostic indicators of survival. „J Clin Oncol”. 24 (27), s. 4464–4471, Sep 2006. DOI: 10.1200/JCO.2006.06.3198. PMID: 16983115.
- ↑ J.H. Lee, R. Essner, H. Torisu-Itakura, L. Wanek i inni. Factors predictive of tumor-positive nonsentinel lymph nodes after tumor-positive sentinel lymph node dissection for melanoma. „J Clin Oncol”. 22 (18), s. 3677–3684, Sep 2004. DOI: 10.1200/JCO.2004.01.012. PMID: 15365064.
- ↑ Coit i in. 2015 ↓, s. 48.
- ↑ a b Coit i in. 2015 ↓, s. 49.
- ↑ a b Coit i in. 2015 ↓, s. 51.
- ↑ a b Coit i in. 2015 ↓, s. 52.
- ↑ Joanna Niesiobędzka-Krężel, Monika Paluszewska. Pegylowane interferony. „Współczesna onkologia”. 5 (4), 2001.
- ↑ J.M. Kirkwood, M.H. Strawderman, M.S. Ernstoff, T.J. Smith i inni. Interferon alfa-2b adjuvant therapy of high-risk resected cutaneous melanoma: the Eastern Cooperative Oncology Group Trial EST 1684. „J Clin Oncol”. 14 (1), s. 7–17, Jan 1996. PMID: 8558223.
- ↑ R.F. Kefford. Adjuvant therapy of cutaneous melanoma: the interferon debate. „Ann Oncol”. 14 (3), s. 358–365, Mar 2003. PMID: 12598338.
- ↑ D.G. Varma, W.R. Richli, C. Charnsangavej, B.I. Samuels i inni. MR appearance of the distended iliopsoas bursa. „AJR Am J Roentgenol”. 156 (5), s. 1025–1028, May 1991. DOI: 10.2214/ajr.156.5.2017926. PMID: 2017926.
- ↑ Dummer i in. 2012 ↓, s. 2.
- ↑ A.M. Eggermont, S. Suciu, M. Santinami, A. Testori i inni. Adjuvant therapy with pegylated interferon alfa-2b versus observation alone in resected stage III melanoma: final results of EORTC 18991, a randomised phase III trial. „Lancet”. 372 (9633), s. 117–126, Jul 2008. DOI: 10.1016/S0140-6736(08)61033-8. PMID: 18620949.
- ↑ a b c d Coit i in. 2015 ↓, s. 50.
- ↑ J.Y. Chen, G. Hruby, R.A. Scolyer, R. Murali i inni. Desmoplastic neurotropic melanoma: a clinicopathologic analysis of 128 cases. „Cancer”. 113 (10), s. 2770–2778, Nov 2008. DOI: 10.1002/cncr.23895. PMID: 18823042.
- ↑ a b Coit i in. 2015 ↓, s. 14.
- ↑ S. Agrawal, J.M. Kane, B.A. Guadagnolo, W.G. Kraybill i inni. The benefits of adjuvant radiation therapy after therapeutic lymphadenectomy for clinically advanced, high-risk, lymph node-metastatic melanoma. „Cancer”. 115 (24), s. 5836–5844, Dec 2009. DOI: 10.1002/cncr.24627. PMID: 19701906.
- ↑ Michael A. Henderson, Bryan Burmeister, Jill Ainslie, Richard Fisher i inni. Adjuvant radiotherapy after lymphadenectomy in melanoma patients: Final results of an intergroup randomized trial (ANZMTG 0.1.02/TROG 02.01).. „Journal of Clinical Oncology”, 2013.
- ↑ a b Coit i in. 2015 ↓, s. 42.
- ↑ B. Zbytek, J.A. Carlson, J. Granese, J. Ross i inni. Current concepts of metastasis in melanoma. „Expert Rev Dermatol”. 3 (5), s. 569–585, Oct 2008. DOI: 10.1586/17469872.3.5.569. PMID: 19649148.
- ↑ a b c d Coit i in. 2015 ↓, s. 53.
- ↑ J.F. Thompson, P.C. Kam. Isolated limb infusion for melanoma: a simple but effective alternative to isolated limb perfusion. „J Surg Oncol”. 88 (1), s. 1–3, Oct 2004. DOI: 10.1002/jso.20112. PMID: 15384062.
- ↑ G.M. Beasley, A. Caudle, R.P. Petersen, N.S. McMahon i inni. A multi-institutional experience of isolated limb infusion: defining response and toxicity in the US. „J Am Coll Surg”. 208 (5), s. 706-715; discussion 715-7, May 2009. DOI: 10.1016/j.jamcollsurg.2008.12.019. PMID: 19476821.
- ↑ C.E. Boesch, T. Meyer, L. Waschke, S. Merkel i inni. Long-term outcome of hyperthermic isolated limb perfusion (HILP) in the treatment of locoregionally metastasised malignant melanoma of the extremities. „Int J Hyperthermia”. 26 (1), s. 16–20, Feb 2010. DOI: 10.3109/02656730903236086. PMID: 20100048.
- ↑ a b Coit i in. 2015 ↓, s. 56.
- ↑ a b c Coit i in. 2015 ↓, s. 60.
- ↑ a b c Coit i in. 2015 ↓, s. 61.
- ↑ a b c d e Coit i in. 2015 ↓, s. 57.
- ↑ a b c Coit i in. 2015 ↓, s. 28.
- ↑ a b c d S. Bhatia, S.S. Tykodi, S.M. Lee, J.A. Thompson. Systemic therapy of metastatic melanoma: on the road to cure. „Oncology (Williston Park)”. 29 (2), s. 126–135, Feb 2015. PMID: 25683834.
- ↑ a b Coit i in. 2015 ↓, s. 58.
- ↑ a b c d F. Spagnolo, P. Ghiorzo, L. Orgiano, L. Pastorino i inni. BRAF-mutant melanoma: treatment approaches, resistance mechanisms, and diagnostic strategies. „Onco Targets Ther”. 8, s. 157–168, 2015. DOI: 10.2147/OTT.S39096. PMID: 25653539.
- ↑ a b c Coit i in. 2015 ↓, s. 33.
- ↑ Coit i in. 2015 ↓, s. 27–28.
- ↑ Coit i in. 2015 ↓, s. 56–57.
- ↑ a b c d P.A. Ascierto, J.M. Kirkwood, J.J. Grob, E. Simeone i inni. The role of BRAF V600 mutation in melanoma. „J Transl Med”. 10, s. 85, 2012. DOI: 10.1186/1479-5876-10-85. PMID: 22554099.
- ↑ H.C. Tang, Y.C. Chen. Insight into molecular dynamics simulation of BRAF(V600E) and potent novel inhibitors for malignant melanoma. „Int J Nanomedicine”. 10, s. 3131–3146, 2015. DOI: 10.2147/IJN.S80150. PMID: 25960652.
- ↑ J.A. Curtin, J. Fridlyand, T. Kageshita, H.N. Patel i inni. Distinct sets of genetic alterations in melanoma. „N Engl J Med”. 353 (20), s. 2135–2147, Nov 2005. DOI: 10.1056/NEJMoa050092. PMID: 16291983.
- ↑ a b K.M. Ilieva, I. Correa, D.H. Josephs, P. Karagiannis i inni. Effects of BRAF mutations and BRAF inhibition on immune responses to melanoma. „Mol Cancer Ther”. 13 (12), s. 2769–2783, Dec 2014. DOI: 10.1158/1535-7163.MCT-14-0290. PMID: 25385327.
- ↑ a b c A.M. Menzies, G.V. Long, R. Murali. Dabrafenib and its potential for the treatment of metastatic melanoma. „Drug Des Devel Ther”. 6, s. 391–405, 2012. DOI: 10.2147/DDDT.S38998. PMID: 23251089.
- ↑ J. Wheler, R. Yelensky, G. Falchook, K.B. Kim i inni. Next generation sequencing of exceptional responders with BRAF-mutant melanoma: implications for sensitivity and resistance. „BMC Cancer”. 15, s. 61, 2015. DOI: 10.1186/s12885-015-1029-z. PMID: 25886620.
- ↑ a b c C. Robert, B. Karaszewska, J. Schachter, P. Rutkowski i inni. Improved overall survival in melanoma with combined dabrafenib and trametinib. „N Engl J Med”. 372 (1), s. 30–39, Jan 2015. DOI: 10.1056/NEJMoa1412690. PMID: 25399551.
- ↑ a b c d G.V. Long, D. Stroyakovskiy, H. Gogas, E. Levchenko i inni. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. „N Engl J Med”. 371 (20), s. 1877–1888, Nov 2014. DOI: 10.1056/NEJMoa1406037. PMID: 25265492.
- ↑ M.M. Chan, L.E. Haydu, A.M. Menzies, M.W. Azer i inni. The nature and management of metastatic melanoma after progression on BRAF inhibitors: effects of extended BRAF inhibition. „Cancer”. 120 (20), s. 3142–3153, Oct 2014. DOI: 10.1002/cncr.28851. PMID: 24985732.
- ↑ M.S. Carlino, K. Gowrishankar, C.A. Saunders, G.M. Pupo i inni. Antiproliferative effects of continued mitogen-activated protein kinase pathway inhibition following acquired resistance to BRAF and/or MEK inhibition in melanoma. „Mol Cancer Ther”. 12 (7), s. 1332–1342, Jul 2013. DOI: 10.1158/1535-7163.MCT-13-0011. PMID: 23645591.
- ↑ a b c d e f A. Niezgoda, P. Niezgoda, R. Czajkowski. Novel Approaches to Treatment of Advanced Melanoma: A Review on Targeted Therapy and Immunotherapy. „Biomed Res Int”. 2015, s. 851387, 2015. DOI: 10.1155/2015/851387. PMID: 26171394.
- ↑ J.A. Sosman, K.B. Kim, L. Schuchter, R. Gonzalez i inni. Survival in BRAF V600-mutant advanced melanoma treated with vemurafenib. „N Engl J Med”. 366 (8), s. 707–714, Feb 2012. DOI: 10.1056/NEJMoa1112302. PMID: 22356324.
- ↑ A. Swaika, J.A. Crozier, R.W. Joseph. Vemurafenib: an evidence-based review of its clinical utility in the treatment of metastatic melanoma. „Drug Des Devel Ther”. 8, s. 775–787, 2014. DOI: 10.2147/DDDT.S31143. PMID: 24966667.
- ↑ P.B. Chapman, A. Hauschild, C. Robert, J.B. Haanen i inni. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. „N Engl J Med”. 364 (26), s. 2507–2516, Jun 2011. DOI: 10.1056/NEJMoa1103782. PMID: 21639808.
- ↑ P.A. Ascierto, D. Minor, A. Ribas, C. Lebbe i inni. Phase II trial (BREAK-2) of the BRAF inhibitor dabrafenib (GSK2118436) in patients with metastatic melanoma. „J Clin Oncol”. 31 (26), s. 3205–3211, Sep 2013. DOI: 10.1200/JCO.2013.49.8691. PMID: 23918947.
- ↑ a b c d e Coit i in. 2015 ↓, s. 34.
- ↑ a b K.T. Flaherty, C. Robert, P. Hersey, P. Nathan i inni. Improved survival with MEK inhibition in BRAF-mutated melanoma. „N Engl J Med”. 367 (2), s. 107–114, Jul 2012. DOI: 10.1056/NEJMoa1203421. PMID: 22663011.
- ↑ C. Robert, R. Dummer, R. Gutzmer, P. Lorigan i inni. Selumetinib plus dacarbazine versus placebo plus dacarbazine as first-line treatment for BRAF-mutant metastatic melanoma: a phase 2 double-blind randomised study. „Lancet Oncol”. 14 (8), s. 733–740, Jul 2013. DOI: 10.1016/S1470-2045(13)70237-7. PMID: 23735514.
- ↑ a b c I.C. Glitza, M.A. Davies. Genotyping of cutaneous melanoma. „Chin Clin Oncol”. 3 (3), s. 27, Sep 2014. DOI: 10.3978/j.issn.2304-3865.2014.03.01. PMID: 25632386.
- ↑ a b M.S. Carlino, J.R. Todd, H. Rizos. Resistance to c-Kit inhibitors in melanoma: insights for future therapies. „Oncoscience”. 1 (6), s. 423–426, 2014. PMID: 25594040.
- ↑ a b J. Guo, L. Si, Y. Kong, K.T. Flaherty i inni. Phase II, open-label, single-arm trial of imatinib mesylate in patients with metastatic melanoma harboring c-Kit mutation or amplification. „J Clin Oncol”. 29 (21), s. 2904–2909, Jul 2011. DOI: 10.1200/JCO.2010.33.9275. PMID: 21690468.
- ↑ D.M. Pardoll. The blockade of immune checkpoints in cancer immunotherapy. „Nat Rev Cancer”. 12 (4), s. 252–264, Apr 2012. DOI: 10.1038/nrc3239. PMID: 22437870.
- ↑ M.A. Postow, M.K. Callahan, J.D. Wolchok. Immune Checkpoint Blockade in Cancer Therapy. „J Clin Oncol”. 33 (17), s. 1974–1982, Jun 2015. DOI: 10.1200/JCO.2014.59.4358. PMID: 25605845.
- ↑ a b c d J.J. Luke, P.A. Ott. PD-1 pathway inhibitors: the next generation of immunotherapy for advanced melanoma. „Oncotarget”. 6 (6), s. 3479–3492, Feb 2015. PMID: 25682878.
- ↑ M. Atefi, E. Avramis, A. Lassen, D.J. Wong i inni. Effects of MAPK and PI3K pathways on PD-L1 expression in melanoma. „Clin Cancer Res”. 20 (13), s. 3446–3457, Jul 2014. DOI: 10.1158/1078-0432.CCR-13-2797. PMID: 24812408.
- ↑ B. Homet Moreno, A. Ribas. Anti-programmed cell death protein-1/ligand-1 therapy in different cancers. „Br J Cancer”. 112 (9), s. 1421–1427, Apr 2015. DOI: 10.1038/bjc.2015.124. PMID: 25856776.
- ↑ O. Hamid, C. Robert, A. Daud, F.S. Hodi i inni. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. „N Engl J Med”. 369 (2), s. 134–144, Jul 2013. DOI: 10.1056/NEJMoa1305133. PMID: 23724846.
- ↑ a b c d T.C. Gangadhar, A.K. Salama. Clinical applications of PD-1-based therapy: a focus on pembrolizumab (MK-3475) in the management of melanoma and other tumor types. „Onco Targets Ther”. 8, s. 929–937, 2015. DOI: 10.2147/OTT.S53164. PMID: 25960664.
- ↑ A. Ribas, I. Puzanov, R. Dummer, D. Schadendorf i inni. Pembrolizumab versus investigator-choice chemotherapy for ipilimumab-refractory melanoma (KEYNOTE-002): a randomised, controlled, phase 2 trial. „Lancet Oncol”, Jun 2015. DOI: 10.1016/S1470-2045(15)00083-2. PMID: 26115796.
- ↑ S.L. Topalian, F.S. Hodi, J.R. Brahmer, S.N. Gettinger i inni. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. „N Engl J Med”. 366 (26), s. 2443–2454, Jun 2012. DOI: 10.1056/NEJMoa1200690. PMID: 22658127.
- ↑ S.L. Topalian, M. Sznol, D.F. McDermott, H.M. Kluger i inni. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. „J Clin Oncol”. 32 (10), s. 1020–1030, Apr 2014. DOI: 10.1200/JCO.2013.53.0105. PMID: 24590637.
- ↑ C. Robert, G.V. Long, B. Brady, C. Dutriaux i inni. Nivolumab in previously untreated melanoma without BRAF mutation. „N Engl J Med”. 372 (4), s. 320–330, Jan 2015. DOI: 10.1056/NEJMoa1412082. PMID: 25399552.
- ↑ J.D. Wolchok, H. Kluger, M.K. Callahan, M.A. Postow i inni. Nivolumab plus ipilimumab in advanced melanoma. „N Engl J Med”. 369 (2), s. 122–133, Jul 2013. DOI: 10.1056/NEJMoa1302369. PMID: 23724867.
- ↑ C. Nakaseko, S. Miyatake, T. Iida, S. Hara i inni. Cytotoxic T lymphocyte antigen 4 (CTLA-4) engagement delivers an inhibitory signal through the membrane-proximal region in the absence of the tyrosine motif in the cytoplasmic tail. „J Exp Med”. 190 (6), s. 765–774, Sep 1999. PMID: 10499915.
- ↑ R.J. Greenwald, M.A. Oosterwegel, D. van der Woude, A. Kubal i inni. CTLA-4 regulates cell cycle progression during a primary immune response. „Eur J Immunol”. 32 (2), s. 366–373, Feb 2002. DOI: 10.1002/1521-4141(200202)32:2<366::AID-IMMU366>3.0.CO;2-5. PMID: 11807776.
- ↑ a b L.H. Camacho. CTLA-4 blockade with ipilimumab: biology, safety, efficacy, and future considerations. „Cancer Med”. 4 (5), s. 661–672, May 2015. DOI: 10.1002/cam4.371. PMID: 25619164.
- ↑ A. Ito, S. Kondo, K. Tada, S. Kitano. Clinical Development of Immune Checkpoint Inhibitors. „Biomed Res Int”. 2015, s. 605478, 2015. DOI: 10.1155/2015/605478. PMID: 26161407.
- ↑ F.S. Hodi, S.J. O’Day, D.F. McDermott, R.W. Weber i inni. Improved survival with ipilimumab in patients with metastatic melanoma. „N Engl J Med”. 363 (8), s. 711–723, Aug 2010. DOI: 10.1056/NEJMoa1003466. PMID: 20525992.
- ↑ C. Robert, L. Thomas, I. Bondarenko, S. O’Day i inni. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. „N Engl J Med”. 364 (26), s. 2517–2526, Jun 2011. DOI: 10.1056/NEJMoa1104621. PMID: 21639810.
- ↑ D. Schadendorf, F.S. Hodi, C. Robert, J.S. Weber i inni. Pooled Analysis of Long-Term Survival Data From Phase II and Phase III Trials of Ipilimumab in Unresectable or Metastatic Melanoma. „J Clin Oncol”. 33 (17), s. 1889–1894, Jun 2015. DOI: 10.1200/JCO.2014.56.2736. PMID: 25667295.
- ↑ A. Ribas, R. Kefford, M.A. Marshall, C.J. Punt i inni. Phase III randomized clinical trial comparing tremelimumab with standard-of-care chemotherapy in patients with advanced melanoma. „J Clin Oncol”. 31 (5), s. 616–622, Feb 2013. DOI: 10.1200/JCO.2012.44.6112. PMID: 23295794.
- ↑ a b c d e f g h i j k l S. Bhatia, S.S. Tykodi, J.A. Thompson. Treatment of metastatic melanoma: an overview. „Oncology (Williston Park)”. 23 (6), s. 488–496, May 2009. PMID: 19544689.
- ↑ M.C. Foletto, S.E. Haas. Cutaneous melanoma: new advances in treatment. „An Bras Dermatol”. 89 (2). s. 301–310. PMID: 24770508.
- ↑ M.B. Atkins, L. Kunkel, M. Sznol, S.A. Rosenberg. High-dose recombinant interleukin-2 therapy in patients with metastatic melanoma: long-term survival update. „Cancer J Sci Am”. 6 Suppl 1, s. S11-4, Feb 2000. PMID: 10685652.
- ↑ M.B. Atkins, M.T. Lotze, J.P. Dutcher, R.I. Fisher i inni. High-dose recombinant interleukin 2 therapy for patients with metastatic melanoma: analysis of 270 patients treated between 1985 and 1993. „J Clin Oncol”. 17 (7), s. 2105–2116, Jul 1999. PMID: 10561265.
- ↑ a b Jacek Mackiewicz, Łukasz Krokowicz. Immunotherapy in advanced cutaneous melanoma patients. „Przegląd Menopauzalny”, 2013. DOI: 10.5114/pm.2013.38599.
- ↑ a b c M.L. Ascierto, I. Melero, P.A. Ascierto. Melanoma: From Incurable Beast to a Curable Bet. The Success of Immunotherapy. „Front Oncol”. 5, s. 152, 2015. DOI: 10.3389/fonc.2015.00152. PMID: 26217587.
- ↑ BioNTech Announces First Patient Dosed in Phase 2 Clinical Trial of mRNA-based BNT111 in Patients with Advanced Melanoma (ang.). Global News Wire, 2021-06-18. [dostęp 2021-06-27].
- ↑ G.V. Long, D. Stroyakovskiy, H. Gogas, E. Levchenko i inni. Overall survival in COMBI-d, a randomized, double-blinded, phase III study comparing the combination of dabrafenib and trametinib with dabrafenib and placebo as first-line therapy in patients (pts) with unresectable or metastatic BRAF V600E/K mutation-positive cutaneous melanoma. „N Engl J Med”. 33 (15), June 2015.
- ↑ a b c P.A. Ascierto, F.M. Marincola, M.B. Atkins. What’s new in melanoma? Combination!. „J Transl Med”. 13, s. 213, 2015. DOI: 10.1186/s12967-015-0582-1. PMID: 26141621.
- ↑ J. Larkin, V. Chiarion-Sileni, R. Gonzalez, J.J. Grob i inni. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. „N Engl J Med”. 373 (1), s. 23–34, Jul 2015. DOI: 10.1056/NEJMoa1504030. PMID: 26027431.
- ↑ DeVita, Lawrence i Rosenberg 2008 ↓, s. 1935.
- ↑ a b c d e Coit i in. 2015 ↓, s. 29.
- ↑ A.Y. Bedikian, M. Millward, H. Pehamberger, R. Conry i inni. Bcl-2 antisense (oblimersen sodium) plus dacarbazine in patients with advanced melanoma: the Oblimersen Melanoma Study Group. „J Clin Oncol”. 24 (29), s. 4738–4745, Oct 2006. DOI: 10.1200/JCO.2006.06.0483. PMID: 16966688.
- ↑ P. Lui, R. Cashin, M. Machado, M. Hemels i inni. Treatments for metastatic melanoma: synthesis of evidence from randomized trials. „Cancer Treat Rev”. 33 (8), s. 665–680, Dec 2007. DOI: 10.1016/j.ctrv.2007.06.004. PMID: 17919823.
- ↑ a b c M.R. Middleton, J.J. Grob, N. Aaronson, G. Fierlbeck i inni. Randomized phase III study of temozolomide versus dacarbazine in the treatment of patients with advanced metastatic malignant melanoma. „J Clin Oncol”. 18 (1), s. 158–166, Jan 2000. PMID: 10623706.
- ↑ C.I. Falkson, J. Ibrahim, J.M. Kirkwood, A.S. Coates i inni. Phase III trial of dacarbazine versus dacarbazine with interferon alpha-2b versus dacarbazine with tamoxifen versus dacarbazine with interferon alpha-2b and tamoxifen in patients with metastatic malignant melanoma: an Eastern Cooperative Oncology Group study. „J Clin Oncol”. 16 (5), s. 1743–1751, May 1998. PMID: 9586887.
- ↑ a b c DeVita, Lawrence i Rosenberg 2008 ↓, s. 1936.
- ↑ P.M. Patel, S. Suciu, L. Mortier, W.H. Kruit i inni. Extended schedule, escalated dose temozolomide versus dacarbazine in stage IV melanoma: final results of a randomised phase III study (EORTC 18032). „Eur J Cancer”. 47 (10), s. 1476–1483, Jul 2011. DOI: 10.1016/j.ejca.2011.04.030. PMID: 21600759.
- ↑ S.S. Legha, S. Ring, A. Bedikian, C. Plager i inni. Treatment of metastatic melanoma with combined chemotherapy containing cisplatin, vinblastine and dacarbazine (CVD) and biotherapy using interleukin-2 and interferon-alpha. „Ann Oncol”. 7 (8), s. 827–835, Oct 1996. PMID: 8922197.
- ↑ M.B. Atkins, J.A. Gollob, J.A. Sosman, D.F. McDermott i inni. A phase II pilot trial of concurrent biochemotherapy with cisplatin, vinblastine, temozolomide, interleukin 2, and IFN-alpha 2B in patients with metastatic melanoma. „Clin Cancer Res”. 8 (10), s. 3075–3081, Oct 2002. PMID: 12374674.
- ↑ P.H. Wiernik, A.I. Einzig. Taxol in malignant melanoma. „J Natl Cancer Inst Monogr”, s. 185–187, 1993. PMID: 7912525.
- ↑ a b R.D. Rao, S.G. Holtan, J.N. Ingle, G.A. Croghan i inni. Combination of paclitaxel and carboplatin as second-line therapy for patients with metastatic melanoma. „Cancer”. 106 (2), s. 375–382, Jan 2006. DOI: 10.1002/cncr.21611. PMID: 16342250.
- ↑ F.E. Nathan, D. Berd, T. Sato, M.J. Mastrangelo. Paclitaxel and tamoxifen: An active regimen for patients with metastatic melanoma. „Cancer”. 88 (1), s. 79–87, Jan 2000. PMID: 10618609.
- ↑ a b A. Pflugfelder, T.K. Eigentler, U. Keim, B. Weide i inni. Effectiveness of carboplatin and paclitaxel as first- and second-line treatment in 61 patients with metastatic melanoma. „PLoS One”. 6 (2), s. e16882, 2011. DOI: 10.1371/journal.pone.0016882. PMID: 21359173.
- ↑ L.M. Evans, E.S. Casper, R. Rosenbluth. Phase II trial of carboplatin in advanced malignant melanoma. „Cancer Treat Rep”. 71 (2), s. 171–172, Feb 1987. PMID: 3542209.
- ↑ D. Khayat, B. Giroux, J. Berille, V. Cour i inni. Fotemustine in the treatment of brain primary tumors and metastases. „Cancer Invest”. 12 (4), s. 414–420, 1994. DOI: 10.3109/07357909409038234. PMID: 8032964.
- ↑ M.F. Avril, S. Aamdal, J.J. Grob, A. Hauschild i inni. Fotemustine compared with dacarbazine in patients with disseminated malignant melanoma: a phase III study. „J Clin Oncol”. 22 (6), s. 1118–1125, Mar 2004. DOI: 10.1200/JCO.2004.04.165. PMID: 15020614.
- ↑ O. Eton, S.S. Legha, A.Y. Bedikian, J.J. Lee i inni. Sequential biochemotherapy versus chemotherapy for metastatic melanoma: results from a phase III randomized trial. „J Clin Oncol”. 20 (8), s. 2045–2052, Apr 2002. PMID: 11956264.
- ↑ M.B. Atkins, J. Hsu, S. Lee, G.I. Cohen i inni. Phase III trial comparing concurrent biochemotherapy with cisplatin, vinblastine, dacarbazine, interleukin-2, and interferon alfa-2b with cisplatin, vinblastine, and dacarbazine alone in patients with metastatic malignant melanoma (E3695): a trial coordinated by the Eastern Cooperative Oncology Group. „J Clin Oncol”. 26 (35), s. 5748–5754, Dec 2008. DOI: 10.1200/JCO.2008.17.5448. PMID: 19001327.
- ↑ N.J. Ives, R.L. Stowe, P. Lorigan, K. Wheatley. Chemotherapy compared with biochemotherapy for the treatment of metastatic melanoma: a meta-analysis of 18 trials involving 2,621 patients. „J Clin Oncol”. 25 (34), s. 5426–5434, Dec 2007. DOI: 10.1200/JCO.2007.12.0253. PMID: 18048825.
- ↑ a b Sławomir Trepka, Piotr Rutkowski, Zbigniew I. Nowecki, Janusz Słuszniak. Chirurgia w leczeniu czerniaków. „Nowotwory”. 60, 2010.
- ↑ a b E. Maverakis, L.A. Cornelius, G.M. Bowen, T. Phan i inni. Metastatic melanoma - a review of current and future treatment options. „Acta Dermato-Venereologica”. 95 (5), s. 516–524, May 2015. DOI: 10.2340/00015555-2035. PMID: 25520039.
- ↑ a b A.M. Leung, D.M. Hari, D.L. Morton. Surgery for distant melanoma metastasis. „The Cancer Journal”. 18 (2). s. 176–184. DOI: 10.1097/PPO.0b013e31824bc981. PMID: 22453019.
- ↑ a b DeVita, Lawrence i Rosenberg 2008 ↓, s. 1932.
- ↑ D.W. Ollila, E.C. Hsueh, S.L. Stern, D.L. Morton. Metastasectomy for recurrent stage IV melanoma. „Journal of Surgical Oncology”. 71 (4), s. 209–213, Aug 1999. PMID: 10440757.
- ↑ Greene i in. 2013 ↓, s. 213.
- ↑ P.V. Dickson, J.E. Gershenwald. Staging and prognosis of cutaneous melanoma. „Surgical Oncology Clinics of North America”. 20 (1), s. 1–17, Jan 2011. DOI: 10.1016/j.soc.2010.09.007. PMID: 21111956.
- ↑ C.C. McLaughlin, X.C. Wu, A. Jemal, H.J. Martin i inni. Incidence of noncutaneous melanomas in the U.S.. „Cancer”. 103 (5), s. 1000–1007, Mar 2005. DOI: 10.1002/cncr.20866. PMID: 15651058.
- ↑ a b c d e f M. Mihajlovic, S. Vlajkovic, P. Jovanovic, V. Stefanovic. Primary mucosal melanomas: a comprehensive review. „International Journal of Clinical and Experimental Pathology”. 5 (8), s. 739–753, 2012. PMID: 23071856.
- ↑ a b c d N. Seetharamu, P.A. Ott, A.C. Pavlick. Mucosal melanomas: a case-based review of the literature. „Oncologist”. 15 (7), s. 772–781, 2010. DOI: 10.1634/theoncologist.2010-0067. PMID: 20571149.
- ↑ a b c d P. Jovanovic, M. Mihajlovic, J. Djordjevic-Jocic, S. Vlajkovic i inni. Ocular melanoma: an overview of the current status. „International Journal of Clinical and Experimental Pathology”. 6 (7), s. 1230–1244, 2013. PMID: 23826405.
- ↑ a b DeVita, Lawrence i Rosenberg 2008 ↓, s. 1952.
- ↑ B.E. Damato, S.E. Coupland. Ocular melanoma. „Saudi Journal of Ophthalmology”. 26 (2), s. 137–144, Apr 2012. DOI: 10.1016/j.sjopt.2012.02.004. PMID: 23960984.
- ↑ a b J.R. Wong, A.A. Nanji, A. Galor, C.L. Karp. Management of conjunctival malignant melanoma: a review and update. „Expert Review of Ophthalmology”. 9 (3), s. 185–204, Jun 2014. DOI: 10.1586/17469899.2014.921119. PMID: 25580155.
- ↑ DeVita, Lawrence i Rosenberg 2008 ↓, s. 1955–56.
- ↑ L. Desjardins, T. Kivelä, B.E. Damato, M.J. Jager: Current Concepts in Uveal Melanoma. Karger Medical and Scientific Publishers, 2011, s. 1. ISBN 978-3-8055-9791-3.
- ↑ a b c d e f P.R. Pereira, A.N. Odashiro, L.A. Lim, C. Miyamoto i inni. Current and emerging treatment options for uveal melanoma. „Journal of Clinical Ophthalmology”. 7, s. 1669–1682, 2013. DOI: 10.2147/OPTH.S28863. PMID: 24003303.
- ↑ I. Puusaari, J. Heikkonen, P. Summanen, A. Tarkkanen i inni. Iodine brachytherapy as an alternative to enucleation for large uveal melanomas. „Ophthalmology”. 110 (11), s. 2223–2234, Nov 2003. DOI: 10.1016/S0161-6420(03)00661-4. PMID: 14597534.
- ↑ B. Damato, I. Patel, I.R. Campbell, H.M. Mayles i inni. Local tumor control after 106Ru brachytherapy of choroidal melanoma. „International Journal of Radiation Oncology”. 63 (2), s. 385–391, Oct 2005. DOI: 10.1016/j.ijrobp.2005.02.017. PMID: 15913907.
- ↑ T.M. Aaberg, C.S. Bergstrom, Z.J. Hickner, M.J. Lynn. Long-term results of primary transpupillary thermal therapy for the treatment of choroidal malignant melanoma. „British Journal of Ophthalmology”. 92 (6), s. 741–746, Jun 2008. DOI: 10.1136/bjo.2007.132951. PMID: 18296506.
- ↑ J.W. Harbour, T.A. Meredith, P.A. Thompson, M.E. Gordon. Transpupillary thermotherapy versus plaque radiotherapy for suspected choroidal melanomas. „Ophthalmology”. 110 (11), s. 2207-2214; discussion 2215, Nov 2003. DOI: 10.1016/S0161-6420(03)00858-3. PMID: 14597531.
- ↑ C.L. Shields, J.A. Shields, N. Perez, A.D. Singh i inni. Primary transpupillary thermotherapy for small choroidal melanoma in 256 consecutive cases: outcomes and limitations. „Ophthalmology”. 109 (2), s. 225–234, Feb 2002. PMID: 11825800.
- ↑ A.A. Yarovoy, D.A. Magaramov, E.S. Bulgakova. The comparison of ruthenium brachytherapy and simultaneous transpupillary thermotherapy of choroidal melanoma with brachytherapy alone. „Brachytherapy”. 11 (3). s. 224–229. DOI: 10.1016/j.brachy.2011.09.007. PMID: 22104351.
- ↑ a b U.B. Hofmann, C.S. Kauczok-Vetter, R. Houben, J.C. Becker. Overexpression of the KIT/SCF in uveal melanoma does not translate into clinical efficacy of imatinib mesylate. „Clinical Cancer Research”. 15 (1), s. 324–329, Jan 2009. DOI: 10.1158/1078-0432.CCR-08-2243. PMID: 19118061.
- ↑ A. Calipel, S. Landreville, A. De La Fouchardière, F. Mascarelli i inni. Mechanisms of resistance to imatinib mesylate in KIT-positive metastatic uveal melanoma. „Clinical and Experimental Metastasis”. 31 (5), s. 553–564, Jun 2014. DOI: 10.1007/s10585-014-9649-2. PMID: 24652072.
- ↑ G.S. Falchook, K.D. Lewis, J.R. Infante, M.S. Gordon i inni. Activity of the oral MEK inhibitor trametinib in patients with advanced melanoma: a phase 1 dose-escalation trial. „The Lancet Oncology”. 13 (8), s. 782–789, Aug 2012. DOI: 10.1016/S1470-2045(12)70269-3. PMID: 22805292.
- ↑ J.M. Kirkwood, L. Bastholt, C. Robert, J. Sosman i inni. Phase II, open-label, randomized trial of the MEK1/2 inhibitor selumetinib as monotherapy versus temozolomide in patients with advanced melanoma. „Clinical Cancer Research”. 18 (2), s. 555–567, Jan 2012. DOI: 10.1158/1078-0432.CCR-11-1491. PMID: 22048237.
- ↑ R. Danielli, R. Ridolfi, V. Chiarion-Sileni, P. Queirolo i inni. Ipilimumab in pretreated patients with metastatic uveal melanoma: safety and clinical efficacy. „Cancer Immunol Immunother”. 61 (1), s. 41–48, Jan 2012. DOI: 10.1007/s00262-011-1089-0. PMID: 21833591.
- ↑ DeVita, Lawrence i Rosenberg 2008 ↓, s. 1920.
- ↑ C. Wei, S. Sirikanjanapong, S. Lieberman, M. Delacure i inni. Primary mucosal melanoma arising from the eustachian tube with CTLA-4, IL-17A, IL-17C, and IL-17E upregulation. „Ear, Nose & Throat Journal”. 92 (1), s. 36–40, Jan 2013. PMID: 23354891.
- ↑ M. Del Vecchio, L. Di Guardo, P.A. Ascierto, A.M. Grimaldi i inni. Efficacy and safety of ipilimumab 3mg/kg in patients with pretreated, metastatic, mucosal melanoma. „European Journal of Cancer”. 50 (1), s. 121–127, Jan 2014. DOI: 10.1016/j.ejca.2013.09.007. PMID: 24100024.
- ↑ J.A. Curtin, K. Busam, D. Pinkel, B.C. Bastian. Somatic activation of KIT in distinct subtypes of melanoma. „Journal of Clinical Oncology”. 24 (26), s. 4340–4346, Sep 2006. DOI: 10.1200/JCO.2006.06.2984. PMID: 16908931.
- ↑ F.S. Hodi, P. Friedlander, C.L. Corless, M.C. Heinrich i inni. Major response to imatinib mesylate in KIT-mutated melanoma. „Journal of Clinical Oncology”. 26 (12), s. 2046–2051, Apr 2008. DOI: 10.1200/JCO.2007.14.0707. PMID: 18421059.
- ↑ J. Lutzky, J. Bauer, B.C. Bastian. Dose-dependent, complete response to imatinib of a metastatic mucosal melanoma with a K642E KIT mutation. „Pigment Cell Melanoma Res”. 21 (4), s. 492–493, Aug 2008. DOI: 10.1111/j.1755-148X.2008.00475.x. PMID: 18510589.
- ↑ X. Jiang, J. Zhou, N.K. Yuen, C.L. Corless i inni. Imatinib targeting of KIT-mutant oncoprotein in melanoma. „Clinical Cancer Research”. 14 (23), s. 7726–7732, Dec 2008. DOI: 10.1158/1078-0432.CCR-08-1144. PMID: 19047099.
- ↑ a b c X. Wang, L. Si, J. Guo. Treatment algorithm of metastatic mucosal melanoma.. „Chinese Clinical Oncology”. 3 (3), s. 38, Sep 2014. DOI: 10.3978/j.issn.2304-3865.2014.08.04. PMID: 25841464.
- ↑ S.N. Markovic, V.J. Suman, J.N. Ingle, J.S. Kaur i inni. Peptide vaccination of patients with metastatic melanoma: improved clinical outcome in patients demonstrating effective immunization. „American Journal of Clinical Oncology”. 29 (4), s. 352–360, Aug 2006. DOI: 10.1097/01.coc.0000217877.78473.a4. PMID: 16891861.
- ↑ L. Si, X. Xu, Y. Kong, K.T. Flaherty i inni. Major response to everolimus in melanoma with acquired imatinib resistance. „J Clin Oncol”. 30 (4), s. e37-40, Feb 2012. DOI: 10.1200/JCO.2011.37.9644. PMID: 22162580.
- ↑ a b B.R. Madewell. Neoplasms in domestic animals: a review of experimental and spontaneous carcinogenesis. „Yale J Biol Med”. 54 (2). s. 111–125. PMID: 7269640.
- ↑ Edward P. Cawley. Genetic aspects of malignant melanoma. „AMA Arch Derm Syphilol.”, 1952. DOI: 10.1001/archderm.1952.01530230064006..
- ↑ C.R. Dorn, W.A. Priester. Epidemiologic analysis of oral and pharyngeal cancer in dogs, cats, horses, and cattle. „J Am Vet Med Assoc”. 169 (11), s. 1202–1206, Dec 1976. PMID: 1002589.
- ↑ E. Kufuor-Mensah, G.L. Watson. Malignant melanomas in a penguin (Eudyptes chrysolophus) and a red-tailed hawk (Buteo jamaicensis). „Vet Pathol”. 29 (4), s. 354–356, Jul 1992. PMID: 1514222.
- ↑ Withrow, Vail i Page 2013 ↓, s. 321.
- ↑ Withrow, Vail i Page 2013 ↓, s. 325–327.
- ↑ William H. Miller Jr., Craig E. Griffin, Karen L. Campbell: Muller and Kirk’s Small Animal Dermatology. Elsevier Health Sciences, 2013, s. 821. ISBN 978-0-323-24193-9.
- ↑ Withrow, Vail i Page 2013 ↓, s. 322–323.
- ↑ a b c d Vito W. Rebecca, Vernon K. Sondak, Keiran S.M. Smalley, A brief history of melanoma: from mummies to mutations, „Melanoma Res”, 22 (2), 2012, s. 114–22, DOI: 10.1097/CMR.0b013e328351fa4d, PMID: 22395415, PMCID: PMC3303163.
- ↑ D.C. Bodenham. A study of 650 observed malignant melanomas in the South-West region. „Ann R Coll Surg Engl”. 43 (4), s. 218–239, Oct 1968. PMID: 5698493. PMCID: PMC2312310.
- ↑ Arvin S. Yang, Paul B. Chapman, The history and future of chemotherapy for melanoma, „Hematology/Oncology Clinics of North America”, 23 (3), 2009, 583-97, x, DOI: 10.1016/j.hoc.2009.03.006, PMID: 19464604, PMCID: PMC3904102.
Bibliografia
- D.G. Coit, J.A. Thompson, R. Andtbacka, C.K. Bichakjian i inni. Melanoma, version 3.2015. „Journal of the National Comprehensive Cancer Network”, 2015.
- D. Weedon i inni, Pathology and Genetics of Tumours of the Skin, IARC Press, 2005, ISBN 92-83-22414-0 [dostęp 2015-09-13] [zarchiwizowane z adresu 2018-07-28].
- Reinhard Dummer, Mark R. Pittelkow, Keiji Iwatsuki, Green Green, Nagwa M. Elwan: Skin Cancer – A World-Wide Perspective: A World-wide Perspective. Springer, 2011. ISBN 978-3-642-05072-5.
- David E. Elder, Sook Jung Yun Yun: Superficial Melanocytic Pathology. Demos Medical Publishing, 2014. ISBN 978-1-62070-023-5.
- Andrzej Szczeklik, Piotr Gajewski: Interna Szczeklika 2014. Kraków: Medycyna Praktyczna, 2014. ISBN 978-83-7430-405-4.
- Guido Massi, Philip E. LeBoit: Histological Diagnosis of Nevi and Melanoma. Springer Science & Business Media, 2013. ISBN 978-3-642-37311-4.
- Walter Mooi, Thomas Krausz: Pathology of Melanocytic Disorders 2ed. CRC Press, 2007. ISBN 978-0-340-80968-6.
- Vinay Kumar, Ramzi S. Cotran, Stanley L. Robins: Robins Patologia. Wrocław: Elsevier Urban & Partner, 2005, s. 232–232. ISBN 83-89581-92-2.
- R. Dummer, A. Hauschild, M. Guggenheim, U. Keilholz i inni. Cutaneous melanoma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. „Annals of Oncology”. 23 Suppl 7, s. vii86-91, Oct 2012. DOI: 10.1093/annonc/mds229. PMID: 22997461.
- Robert Johr, Wilhelm Stolz: Dermoscopy: An Illustrated Self-Assessment Guide: An Illustrated Self-Assessment Guide Ebook. McGraw Hill Professional, 2010. ISBN 978-0-07-161356-9.
- Vincent T. DeVita, Theodore S. Lawrence, Steven A. Rosenberg: Devita, Hellman & Rosenberg’s Cancer: Principles & Practice of Oncology. Wyd. 8. Lippincott Williams & Wilkins, 2008. ISBN 978-0-7817-7207-5.
- Dennis A. Casciato, Territo Mary C.: Manual of Clinical Oncology. Lippincott Williams & Wilkins, 2012. ISBN 978-1-4511-1560-4.
- Dawid Morris: Cancer. CRC Press, 2003. ISBN 978-0-203-30416-7.
- Frederick L. Greene, David L. Page, Irvin D. Fleming, April G. Fritz, Charles M. Balch, Daniel G. Haller, Monica Morrow: AJCC Cancer Staging Manual. Springer Science & Business Media, 2013. ISBN 978-1-4757-3656-4.
- Stephen J. Withrow, David M. Vail, Rodney Page: Withrow and MacEwen’s Small Animal Clinical Oncology. Elsevier Health Sciences, 2013. ISBN 978-0-323-24197-7.
![]() Przeczytaj ostrzeżenie dotyczące informacji medycznych i pokrewnych zamieszczonych w Wikipedii.
Przeczytaj ostrzeżenie dotyczące informacji medycznych i pokrewnych zamieszczonych w Wikipedii.
Media użyte na tej stronie

The Star of Life, medical symbol used on some ambulances.
Star of Life was designed/created by a National Highway Traffic Safety Administration (US Gov) employee and is thus in the public domain.Autor: LWozniak&KWZielinski, Licencja: CC BY-SA 3.0
Czerniak, postać guzkowa
- Nowotwór rozrasta się nie tylko wgłębnie (wertykalnie), ale z jednego boku (lewa strona mikrofotografii) także powierzchniowo (radialnie). Melanina jest rozmieszczona nierównomiernie.
Autor: Emmanouil K Symvoulakis, Dionysios E Kyrmizakis, Emmanouil I Drivas, Anastassios V Koutsopoulos, Stylianos G Malandrakis, Charalambos E Skoulakis and John G Bizakis, Licencja: CC BY-SA 2.0
Photograph at initial examination showing an oversized pigmented soft mass (melanoma) arising from the palate.
Autor: S. Sharma , N.L. Sharma, V. Sharma, Licencja: GFDL
5-day-old girl (head to right) with giant congenital melanocytic nevus over back, neck, buttocks and left thigh. Histopathological findings showed nevomelanocytes throughout the thickness of the dermis and extending into the subcutaneous tissue. No other complications were reported.
Title: Pathology: Patient: Melanoma
Description: This is advanced malignant melanoma. At the left, one can see a plaque of early, radial growth phase superficial spreading melanoma. To the right, and contiguous with the plaque, is a pink (amelanotic) nodule of deeply invasive vertical growth phase melanoma. Melanomas diagnosed at this stage have a poor prognosis; many of these patients develop metastatic disease and die from their cancer. In the majority of instances, the plaque stage of melanoma is present for a sufficient period of time to permit its diagnosis and removal before it progresses to a more advanced (and more difficult to treat) stage.
Topics/Categories: Pathology -- Patient
Type: Color Slide
Source: National Cancer Institute
Author: Unknown photographer/artist
AV Number: AV-8500-3606
Date Created: 1985
Date Entered: 1/1/2001
Autor: Ed Uthman from Houston, TX, USA, Licencja: CC BY 2.0
Native liver tissue is strongly positive, while the tumor cells are faintly positive. CAM5.2 can be positive in up to 10% of melanomas. Cytokeratins AE1-AE3 were negative in this case.
Six bottles of different types of cancer drugs over a graded blue to white background. Clockwise from center: Blenoxane (bleomycin), Oncovin (vincristine), DTIC-Dome (dacarbazine), Cytoxan (cyclophosphamide), Adriamycin (doxorubicin), and VePesid (etoposide).
Autor: Dr Laughlin Dawes, Licencja: CC BY 3.0
"This 59 year-old female patient presented with acute right hemiplegia, aphasia and confusion. She had a known cerebral melanoma metastasis in the left frontal lobe. This axial T2-weighted MR (click image for arrows) shows a large haematoma with a fluid-fluid level (green arrows). There is a smaller low signal area anteriorly (red arrows) which corresponded to the known metastasis. This smaller area enhanced after gadolinium, as did the overlying dura." (dr Dawes)
- Title Interferon Bottles
- Description Seen are three bottles of interferon on a black background.
- Topics/Categories Treatment, Chemotherapy
- Type Color, Photo
- Source National Cancer Institute
Autor: Dfvchrome, Licencja: CC0
Sun Blisters on a shoulder after 48 hours of exposure to sun.
- Title Dysplastic Nevi
- Description This lesion has a dark brown, "pebbly" elevated surface against a lighter tan, macular background. The irregular, indistinct margin of the nevus helps to distinguish it from the small congenital-pattern nevus, which some dysplastic nevi closely resemble clinically. Its distinctive morphology, not its size (6 by 6 mm), identifies it as a dysplastic nevus. For additional resource, see the following web site: http://www.cancer.gov/cancertopics/wyntk/melanoma See also http://www.cancer.gov/cancertopics/wyntk/moles-and-dysplastic-nevi.
- Topics/Categories Anatomy -- Skin Cancer Types -- Skin Cancer Cells or Tissue -- Abnormal Cells or Tissue
- Type Color, Photo
- Source National Cancer Institute
Autor:
- Fæ
- Cancer_Research_UK_uploader
- derivative work: M1llx (zgłoś błąd)
Izolowana infuzja do kończyny
Autor: Jonathan Trobe, M.D. - University of Michigan Kellogg Eye Center, Licencja: CC BY 3.0
Photograph of an iris melanoma
This image depicts a patient’s chest, which displays numerous darkly-pigmented cutaneous lesions, which had been diagnosed as malignant melanoma (MM).
When diagnosing MM from a gross pathologic perspective, i.e., with the naked eye, a physician must keep in mind the “ABCDE” rule, which represents:
- A = Asymmetry – Is the shape of the lesion(s) symmetrical, evenly distributed/shaped?
- B = Border Irregularity – Is the border of the lesion(s) smooth or rough?
- C = Color Variability – Is the lesion(s) multicolored, or of a single color?
- D = Diameter – Is the lesion(s) greater than 6mm (0.24in.)?
- E = Evolution – How did the lesion(s) evolve over time, i.e., did it appear, or change, quickly, or over a long
period of time?Autor: Nephron, Licencja: CC BY-SA 3.0
High magnification micrograph of lentigo maligna, also known as melanoma in situ with solar damage. H&E stain.
Lentigo maligna should not be confused with lentigo maligna melanoma (LMM). LMM is a melanocytic lesion that has a dermal component.[1]
Related images

Intermed. mag.
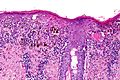
High mag.
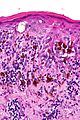
Very high mag.



References
- ↑ (Apr 2006). "Lentigo maligna/lentigo maligna melanoma: current state of diagnosis and treatment.". Dermatol Surg 32 (4): 493-504. DOI:10.1111/j.1524-4725.2006.32102.x.
Autor: LWozniak&KWZielinski, Licencja: CC BY-SA 3.0
Czerniak dystalny
- Rozrost atypowych melanocytów, głównie na granicy skórno-naskórkowej. W skórze nacieki zapalne, naskórek nadmiernie zrogowaciały.
Autor: Hellerhoff, Licencja: CC BY-SA 4.0
Aderhautmelanom in der Kernspintomographie T2 axial
Autor: Department of Pathology, Calicut Medical College, Licencja: CC BY-SA 4.0
Gross appearance of lymphnode metastasis from melanoma
Autor: Ed Uthman from Houston, TX, USA, Licencja: CC BY 2.0
This tumor grew back, despite two previous attempts at removal. Melanoma is caused by exposure to sunlight and tanning booth light. One or more severe sunburns in your teens and twenties are especially harmful.
Autor: 0x6adb015, Licencja: CC BY-SA 3.0
Someone with Dysplastic Nevus Syndrome
René-Théophile-Hyacinthe Laennec (1781 – 1826) French physician and inventor of the stethoscope.

Autor: Ja, właściciel praw autorskich do tego dzieła, udostępniam je na poniższej licencji, Licencja: CC BY-SA 3.0
Endoscopic view of a dark gray polypoidal tumor of the esophagus at presentation.
Autor: LWozniak&KWZielinski, Licencja: CC BY-SA 3.0
Czerniak desmoplastyczny
- Powierzchnia guza owrzodziała. Pod strupem (górna część obrazu) widoczne są wydłużone komórki o różnokierunkowym ułożeniu.
Autor: LWozniak&KWZielinski, Licencja: CC BY-SA 3.0
Czerniak na podłożu znamienia błękitnego
- Wrzecionowate melanocyty o pasmowatym układzie zawieraja sporo melaniny. O rozpoznaniu zadecydował stopień atypii komórkowej, duży stosunek jądrowo-cytoplazmatyczny oraz duża liczba mitoz, obecność atypowych figur podziałowych (np. w środku ryciny).
Metastatic Melanoma in Lymph Node
This 6.5-centimeter mass was removed from the axilla of a middle-aged woman, who was a longtime resident of a developmental center for the severely disabled. She was unable to give any history, but there was no medical record of a primary melanoma, and we were unable to find any biopsy scars at the time of surgery. The dark brown color of the cut surface provided a clue to the diagnosis, an unusual finding in metastatic melanomas, which are more typically amelanotic (like the upper right segment of this specimen). Pigment was easily demonstrated in the tumor cells at frozen section, allowing a definitive diagnosis intraoperatively.
Photograph by Ed Uthman, MD. Public domain. Posted 30 May 99- Title Melanoma with Diameter Change
- Description Seen is melanoma with the diameter that had changed in size. Part of the ABCDs for detection of melanoma. For additional resource, see the following web site: http://www.cancer.gov/cancertopics/wyntk/moles-and-dysplastic-nevi See also http://www.cancer.gov/cancertopics/wyntk/melanoma.
- Topics/Categories Anatomy -- Skin Cancer Types -- Melanoma Cancer Types -- Skin Cancer Cells or Tissue -- Abnormal Cells or Tissue
- Type Color, Photo
- Source Skin Cancer Foundation
This slide shows a melanoma on a patient's skin.
- Title Melanoma
- Description This is advanced malignant melanoma. At the left, one can see a plaque of early, radial growth phase superficial spreading melanoma. To the right, and contiguous with the plaque, is a pink (amelanotic) nodule of deeply invasive vertical growth phase melanoma. Melanomas diagnosed at this stage have a poor prognosis; many of these patients develop metastatic disease and die from their cancer. In the majority of instances, the plaque stage of melanoma is present for a sufficient period of time to permit its diagnosis and removal before it progresses to a more advanced (and more difficult to treat) stage. See also http://www.cancer.gov/cancertopics/wyntk/moles-and-dysplastic-nevi.
- Topics/Categories Anatomy -- Skin Cancer Types -- Melanoma Cancer Types -- Skin Cancer Cells or Tissue -- Abnormal Cells or Tissue
- Type Color, Photo
- Source National Cancer Institute
HEART-GREAT VESSELS: METASTATIC MALIGNANT MELANOMA: HEART
Note the pigmented masses throughout the heart.- Title Dysplastic Nevi
- Description The central portion of this mole is a complex papule. The periphery of the lesion is macular, irregular, indistinct and slightly pink. For additional resource, see the following web site: http://www.cancer.gov/cancertopics/wyntk/skin See also http://www.cancer.gov/cancertopics/wyntk/moles-and-dysplastic-nevi..
- Topics/Categories Anatomy -- Skin Cancer Types -- Skin Cancer Cells or Tissue -- Abnormal Cells or Tissue
- Type Color, Photo
- Source National Cancer Institute
Autor: Autor nie został podany w rozpoznawalny automatycznie sposób. Założono, że to KGH (w oparciu o szablon praw autorskich)., Licencja: CC-BY-SA-3.0
Histopathologic image of malignant melanoma (Case 01). Skin biopsy. The same case as illustrated in a file "Malignant_melanoma_(1)_at_thigh_Case_01.jpg". H & E stain. This case may represent superficial spreading melanoma[1]
- Title Melanoma, Brown Lesion
- Description A brown lesion with a large and irregular border on the skin. Melanoma with characteristic asymmetry, border irregularity, color variation, and large diameter.
- Topics/Categories Anatomy -- Skin Cancer Types -- Melanoma Cancer Types -- Skin Cancer Cells or Tissue -- Abnormal Cells or Tissue
- Type Color, Photo
- Source National Cancer Institute
- Title Dysplastic Nevi
- Description This large (7 by 11 mm) macular lesion displays an irregular, scalloped border, which is indistinct in some areas. In addition to hues of tan and brown, several pink areas (arrows) are present. The presence of pink colors in the macular portion of a melanocytic nevus is quite distinctive for dysplastic nevi. For additional resource, see the following web site: http://www.cancer.gov/cancertopics/wyntk/skin See also http://www.cancer.gov/cancertopics/wyntk/moles-and-dysplastic-nevi.
- Topics/Categories Anatomy -- Skin Cancer Types -- Skin Cancer Cells or Tissue -- Abnormal Cells or Tissue
- Type Color, Photo
- Source National Cancer Institute