Heteroazeotrop
Heteroazeotrop – trójfazowa mieszanina azeotropowa, czyli taki azeotrop, w którym z parą współistnieją różne niemieszające się roztwory, np. dwie fazy ciekłe (w przypadku układów dwuskładnikowych). Na wykresach fazowych odniesionych do stałego ciśnienia znajduje się odcinek izotermy, którego końce określają zawartości składników w obu współistniejących roztworach (roztwory wzajemnie nasycone). Punkt określający skład pary leży pomiędzy nimi. Skład pary nie zależy od udziałów obu faz ciekłych w układzie – nie ulega zmianom dopóki w układzie znajdują się oba roztwory, co jest konsekwencją reguły faz Gibbsa[1][2][3].
Podstawy fizykochemiczne
Azeotropy powstają w sytuacji, gdy roztwór rzeczywisty wykazuje znaczne odstępstwa od prawa Raoulta. Duże odstępstwa dodatnie występują wówczas, gdy prężność pary nad mieszaniną A i B jest większa od prężności obliczonej przy założeniu addytywności (prawo Raoulta). Może być większa od prężności par nad czystymi składnikami A i B. Przyczyną jest dużo większe przyciąganie międzycząsteczkowe A–A i B–B od przyciągania A–B. W tej sytuacji temperatura wrzenia roztworu dwuskładnikowego jest niższa od temperatur wrzenia składników. Przy określonej proporcji A : B występuje minimum temperatury wrzenia (maksimum prężności par – azeotrop dodatni).
Różnica między siłami oddziaływania między cząsteczkami jednakowymi i różnymi może się zwiększać w miarę ochładzania roztworu. W niektórych wypadkach prowadzi to do pojawienia się zmętnienia, a następnie rozwarstwienia cieczy. Powstają dwa współistniejące roztwory nasycone (A w B i B w A). Zakres ograniczonej mieszalności cieczy przeważnie rozszerza się w miarę dalszego obniżania temperatury.
Na wykresach fazowych ilustrujących heteroazeotropię obszary współistnienia cieczy z parą i obszar współistnienia dwóch cieczy są połączone. Granicą między nimi jest odcinek izotermy, na którym mieszczą się punkty odpowiadające wszystkim układom trójfazowym ciecz–ciecz–para. Zgodnie z regułą faz Gibbsa takie układy są inwariantne (niezmiennicze, zerozmienne) – nie dysponują żadnym stopniem swobody (s = 0). Pozostają w stałej temperaturze tak długo, dopóki nie zmniejszy się liczba współistniejących faz.

Azeotrop dodatni i obszar ograniczonej mieszalności cieczy
1 – para, 2 – roztwór AB, 2′ – nasycony roztwór B w A, 2″ – nasycony roztwór A w B, 1+2, 2′+2″ – obszary współistnienia dwóch faz, a, m, b – sposób określania ilości współistniejących faz, a i b na podstawie reguły dźwigni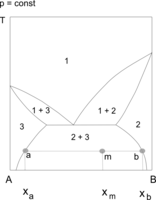
Heteroazeotropia w układzie dwuskładnikowym (AB)
1 – para, 2 – roztwór A w B, 3 – roztwór B w A, 1+2, 1+3, 2+3 – obszary współistnienia dwóch faz, a, m, b – sposób określania ilości współistniejących faz na podstawie reguły dźwigni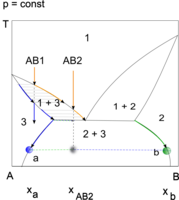
Przykład interpretacji wykresu fazowego. Proces chłodzenia pary AB1 i AB2
linia pomarańczowa – para (1), linia granatowa – roztwór B w A (3), linia zielona – roztwór A w B (2); stosunek długości zielonej i granatowej linii przerywanej w obszarze „2 + 3” odpowiada stosunkowi ilości obu roztworów
Przebieg kondensacji heteroazeotropu
Ochładzanie par zawierających składniki A i B w ilościach odpowiadających heteroazeotropowi przebiega swobodnie aż do osiągnięcia temperatury przemiany heteroazeotropowej. W tej temperaturze rozpoczyna się równoczesna kondensacja nie mieszających się wzajemnie nasyconych roztworów B(A) i A(B) (w nawiasach – substancja rozpuszczona). Temperatura pozostaje stała do zakończenia kondensacji (s = 0).
Dwufazowy kondensat (złożony np. z warstwy wodnej i hydrofobowej warstwy organicznej) może być rozdzielony bezpośrednio po skropleniu par lub po dalszym chłodzeniu, które może powodować zmiany składu obu cieczy i ich udziałów. Zmiany udziałów występują wtedy, gdy różny jest kąt nachylenia obu granic obszaru dwufazowego, który ilustruje zależność rozpuszczalności od temperatury. W przypadku dużej asymetrii obszaru niemieszalności chłodzenie powoduje wyraźną zmianę stosunku ilości faz (różne przyrosty ramion „dźwigni”).
Jeżeli para A + B zawiera nadmiar składnika A w stosunku do składu azeotropu, jej skraplanie rozpoczyna się w temperaturze, w której jest osiągana górna granica obszaru współistnienia pary z roztworem A(B). Skład pierwszej kropli kondensatu określa punkt przecięcia izotermy z dolną granicą tego obszaru. Dalszy przebieg kondensacji ilustruje droga trzech punktów, opisujących:
- skład układu ciecz–para (punkt przemieszcza się pionowo w dół),
- skład pary (punkt przemieszcza się w kierunku mniejszych stężeń A, wzdłuż górnej granicy obszaru dwufazowego),
- skład cieczy (punkt przemieszcza się w kierunku mniejszych stężeń A, wzdłuż dolnej granicy obszaru dwufazowego).
Ilość pary i cieczy w dowolnej temperaturze określa się na podstawie reguły dźwigni.
Kondensacja kończy się przed osiągnięciem temperatury przemiany azeotropowej w tych przypadkach, gdy stężenie B w parze jest mniejsze od granicznej rozpuszczalności B w A. Temperaturę zakończenia kondensacji wskazuje wówczas punkt przecięcia dolnej granicy obszaru ciecz–para z linią składu układu AB.
W przypadku większych stężeń składnika B kondensacja kończy się w temperaturze azeotropowej. Ta temperatura utrzymuje się od chwili zmętnienia kondensatu A(B), spowodowanego powstaniem pierwszych kropli roztworu B(A), do zaniku pary. Dwufazowy kondensat może być odbierany osobno, po odebraniu frakcji jednofazowej, po czym rozdzielany, np. w celu wyodrębnienia roztworu bogatego w składnik B. Ilość kondensatu dwufazowego zależy od tego, jaka jest różnica między początkowym składem pary i składem azeotropu.
W przypadkach, gdy stężenie B w parze jest większe od stężenia azeotropowego kondensacja przebiega analogicznie. Kondensat dwufazowy może być odbierany po odebraniu frakcji jednofazowej – roztworu B(A).
Zobacz też
Przypisy
- ↑ Stanisław Bursa: Chemia fizyczna. Wyd. Wyd. 2 popr. Warszawa: Państwowe Wydawnictwo Naukowe, 1979. ISBN 83-01-00152-6. (pol.).
- ↑ Janusz Ciborowski: Podstawy inżynierii chemicznej. Wyd. Wyd. 1. Warszawa: Wydawnictwa Naukowo-Techniczne, 1965. (pol.).
- ↑ Antoni Basiński, Adam Bielański, Kazimierz Gumiński, i inni: Chemie fizyczna. Wyd. Wyd. 3. Warszawa: Państwowe Wydawnictwo Naukowe, 1966. (pol.).
Media użyte na tej stronie

Autor: Joanna Kośmider, Licencja: CC BY 3.0
Wykresy fazowe/równowaga ciecz-ciecz 4b

Autor: Joanna Kośmider, Licencja: CC BY 3.0
Phase diagrams/ liquid-liquid equilibrium 4

Autor: Joanna Kośmider, Licencja: CC BY 3.0
Phase diagrams/ liquid-liquid equilibrium 3