Metoda Sangera
Metoda Sangera, metoda dideoksy – jeden ze sposobów sekwencjonowania DNA, stanowiący (z różnymi modyfikacjami) podstawę odczytywania sekwencji DNA, polegający na kopiowaniu, katalizowanym przez polimerazę DNA, badanej cząsteczki DNA w warunkach in vitro.
Za opracowanie tej metody Frederick Sanger otrzymał w 1980 roku nagrodę Nobla[1].
Opis metody
Przebieg reakcji można podzielić na pięć etapów:
- preparatywny PCR – otrzymanie homogennego DNA
- denaturacja – otrzymanie jednoniciowej matrycy
- synteza nowych nici z użyciem dNTP i ddNTP oraz startera
- elektroforeza na żelu poliakrylamidowym ze stałym stężeniem środka denaturującego
- analiza prążków.
Reakcja przeprowadzana jest w czterech wersjach (próbówkach), w każdej z nich przy wykorzystaniu próbki tego samego badanego DNA, ale różnych odczynników. W jednym z wariantów reakcja przeprowadzana jest tak, że wszystkie jej produkty zakończone są nukleotydem adeninowym. Powstają cząsteczki DNA, których sekwencja jest wprawdzie jeszcze nieznana, ale wiadomo, że są komplementarnymi kopiami pewnych fragmentów badanego DNA, a ich ostatnim nukleotydem jest nukleotyd adeninowy. Zgodnie z zasadą komplementarności adenina ma w matrycowym DNA swój odpowiednik w formie nukleotydu tyminowego. Ponieważ w naturalnych cząsteczkach DNA każdy z czterech rodzajów nukleotydów występuje wiele razy, to fragmenty zakończone jednego rodzaju nukleotydem mogą mieć bardzo różną długość. Wszystkie jednak fragmenty i to we wszystkich wariantach doświadczenia, zaczynają się takim samym odcinkiem - starterem – dodanym na początku reakcji wraz z pozostałymi substratami. Podobnie w pozostałych wariantach reakcji otrzymuje się produkty mające różną długość, lecz zawierające starter na jednym końcu (końcu 5'), a jednego rodzaju nukleotyd na drugim końcu (końcu 3'). Cztery możliwe warianty reakcji sekwencjonowania to wariant A - prowadzący do otrzymania różnej długości produktów zawsze zakończonych nukleotydem adeninowym, C - wszystkie produkty zakończone nukleotydem cytozynowym i analogicznie warianty dla T i G.

Podczas kopiowania jednej cząsteczki DNA o tym, kiedy nastąpi przerwanie syntezy DNA, decyduje przypadek. Ponieważ każda z czterech próbówek zawiera miliony cząsteczek analizowanego DNA i każda z tych cząsteczek ulega kopiowaniu, to zgodnie z prawem wielkich liczb w próbówkach tych pojawiają się łańcuchy o wszelkich możliwych długościach (zależnych od sekwencji matrycy). Na przykład jeśli cząsteczka badanego DNA ma następującą postać: [odcinek komplementarny do startera]GATTCGA, to w reakcji typu A mogą powstać następujące trzy rodzaje produktów:
- [starter]CTA
- [starter]CTAA
- [starter]CTAAGCT
DNA matrycowy (badany):
- [odcinek komplementarny do startera]GATTCGA.

Jak widać, od reguły, że w reakcji A wszystkie produkty zakończone są nukleotydem adeninowym może zdarzyć się wyjątek, ale dotyczy to tylko najdłuższych produktów, łatwych do rozpoznania (najdłuższe produkty pomija się w analizie). W próbówce wszystkie te cząsteczki są wymieszane, można je jednak uporządkować pod względem wielkości, poddając roztwory otrzymane po reakcji sekwencjonowania rozdziałowi elektroforetycznemu. Sanger zastosował do tego wysokorozdzielczą elektroforezę żelową, umożliwiającą rozróżnienie nawet łańcuchów DNA różniących się tylko jednym nukleotydem. Im mniejsze są cząsteczki DNA, tym dalej wędrują podczas doświadczenia od ujemnej elektrody. Produkty czterech wariantów reakcji sekwencjonowania nanosi się na żel tuż obok siebie, aby łatwiej było je porównywać. Po przeprowadzeniu rozdziału cząsteczek DNA trzeba je uwidocznić. Do niedawna standardowym podejściem do tego było takie przeprowadzanie reakcji sekwencjonowania, aby jej produkty były radioaktywne (znakowane izotopowo); w takim przypadku do żelu elektroforetycznego należy przyłożyć kliszę fotograficzną (rentgenowską) i przez kilkadziesiąt godzin przeprowadzać jej naświetlanie. Ostatecznie uzyskuje się autoradiogram - obraz różnych frakcji DNA w formie ciemnych prążków na wywołanej kliszy. Najmniejszym cząsteczkom odpowiadają prążki położone najdalej od jednej z krawędzi kliszy (odpowiadającej miejscu naniesienia próbek na żel). Pozostaje wtedy przeprowadzenie analizy układu frakcji DNA. W przypadku sekwencjonowania powyższego przykładowego DNA zobaczylibyśmy gdzieś na autoradiogramie prążek odpowiadający frakcji złożonej z bardzo niewielkich, szybko wędrujących cząsteczek skopiowanego DNA. Powiedzmy, że dostrzeżemy go w linii rozdziału produktów reakcji typu G. Będzie to oznaczać, że produkty sekwencjonowania zawierają nukleotyd guaninowy gdzieś w niewielkiej odległości od startera. Powinniśmy wtedy odszukać na autoradiogramie prążek odpowiadający fragmentom DNA minimalnie dłuższym od analizowanych poprzednio (minimalnie dłuższe to dłuższe o jeden nukleotyd). Przy użyciu podanej tu przykładowej sekwencji taki minimalnie dłuższy fragment zostałby znaleziony wśród produktów reakcji typu A. Kojarząc wnioski z analizy obu sąsiednich prążków dojdziemy do wniosku, że produkty sekwencjonowania w pewnej niewielkiej odległości od startera zawierają kolejno nukleotydy G i A. Podobnie następne prążki (pod względem odległości od linii naniesienia prób) zobaczylibyśmy wśród produktów reakcji typu T, znowu wśród produktów reakcji T, potem reakcji typu A, C, G, T itd. W ten sposób uzyskujemy sekwencję nici DNA komplementarnej wobec tej, która była kopiowana w reakcji sekwencjonowania, odczytujemy kolejno nukleotydy coraz dalsze od startera. Ponieważ DNA składa się z dwóch wzajemnie komplementarnych wobec siebie nici, jest to zarazem gotowa sekwencja tej drugiej nici, komplementarnej wobec skopiowanej matrycy.


Jak wynika z powyższego, warunkiem odczytania sekwencji DNA, być może dość długiej, liczącej np. 400 - 600 nukleotydów jest wcześniejsze poznanie niewielkiego jej fragmentu (w idealnych warunkach mającego długość około 20 nukleotydów), tak by można było przygotować starter odpowiedni do tej matrycy. Problem ten jest często rozwiązywany w ten sposób, że badane, nieznane DNA wbudowuje się (przy pomocy enzymu ligazy) do pomocniczej cząsteczki DNA o znanej sekwencji, zwanej wektorem klonującym czy wektorem do klonowania DNA (genów). Przystępując do sekwencjonowania, dobiera się starter komplementarny wobec fragmentu wektora (znanego) sąsiadującego z nieznanym, badanym fragmentem DNA.
Istotnym elementem metody Sangera jest otrzymywanie populacji cząsteczek DNA będących komplementarnymi kopiami matrycy i zakończonych jednego rodzaju nukleotydem. Powielanie cząsteczki DNA matrycowego (badanego) przeprowadza się w mieszaninach reakcyjnych zawierających wszystkie substraty niezbędne dla polimerazy DNA i jeszcze jeden substrat nietypowy. Składnikami typowymi, identycznymi we wszystkich wariantach tego doświadczenia, są (poza samym matrycowym DNA i enzymem): startery, dATP, dCTP, dGTP, dTTP (2'-deoksyadenozynotrójfosforan itp., ogólnie dNTP), bufor nadający roztworowi odpowiednie pH i zawierający odpowiednie jony aktywatory - jony Mg2+. Ponadto jedna z próbówek (wariant A) zawiera nietypowy nukleotyd ddATP (2',3'-dideoksyadenozynotrójfosforan). Kiedy polimeraza DNA odczytuje w matrycy nukleotyd tyminowy, to w nowo syntetyzowanej nici wbudowuje komplementarny nukleotyd adeninowy wykorzystując dATP jako substrat. Ponieważ w roztworze znajduje się także analog dideksy, ddATP, a polimeraza nie rozróżnia tych dwóch związków, do niektórych nowych łańcuchów DNA wbudowany zostaje nukleotyd zmodyfikowany. O tym który z tych nukleotydów zostanie przez enzym wykorzystany na danym etapie kopiowania decyduje przypadek. Jeśli przyłączony zostanie naturalny nukleotyd adeninowy, to polimeraza może kontynuować syntezę DNA. Jeśli wbudowana zostanie dideoksyadenozynofosforan, to proces syntezy zostaje przerwany, gdyż analog ten nie ma w pozycji 3' grupy hydroksylowej umożliwiającej przyłączenie kolejnego nukleotydu. Podobnie mieszanina reakcyjna typu C zawiera poza dNTP, także ddCTP, mieszanina typu G - dNTP i ddGTP, a roztwór typu T - dNTP i ddTTP, a synteza łańcuchów DNA w tych mieszaninach przerwana gdy polimeraza wbuduje do niego dideoksynukleotyd.
- Terminacja syntezy DNA przez dideoksynukleozyd włączony do rosnącego łańcucha
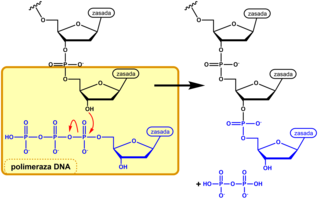
Przyłączenie nukleotydu przez polimerazę do rosnącej naturalnej nici DNA

Terminacja elongacji DNA przez dideoksynukleozyd
- Modyfikacje metody Sangera
- sekwencjonowanie cykliczne
- znakowanie fluorescencyjne.
Zobacz też
- metoda Maxama-Gilberta
Przypisy
- ↑ Sanger, F, Nicklen, S, Coulson, AR. DNA sequencing with chain-terminating inhibitors. „Proceedings of the National Academy of Sciences of the United States of America”. 74. 12, s. 5463–5467, 1977. PMID: 431765.
Bibliografia
- Robert K. Murray (red.): Biochemia Harpera. Warszawa: PZWL, 2005. ISBN 83-200-3347-0.
Media użyte na tej stronie

Autor: Christoph Goemans (modifiziert), Licencja: CC BY-SA 3.0
Prinzip der DNA-Sequenzierung nach der Didesoxy-Methode. dNTP ist die allgemeine Abkürzung für ein Nucleotid und kann für dATP, dCTP, dGTP oder dTTP stehen. ddNTPs sind die entsprechenden Didesoxy-Varianten der dNTPs. Der Einbau eines ddNTPs führt zum Abbruch der Polymerisationsreaktion. Die blauen Punkte am 5'-Ende des Primers stellen eine Markierung dar (z.B. eine fluoreszierende Gruppe), mittels der die Syntheseprodukte später im Gel sichtbar gemacht werden können. Alternativ lassen sich auch radioaktiv markierte Nucleotide zur Polymerisationsreaktion einsetzen.
Structure of dideoxyadenosine triphosphate; 2',3'-dideoxyadenosine triphosphate; ddATP; 2',3'-Dideoxy-ATP
Structure of Deoxyadenosine triphosphate
Autor: John Schmidt, Licencja: CC-BY-SA-3.0
DNA sequencing gel. Sequence visualized by autoradiography.

Autor: Michał Sobkowski, Licencja: CC BY 3.0
Terminacja elongacji DNA przez nukleozyd dideoksy