Metylacja DNA
| Ten artykuł należy dopracować |
Metylacja DNA – proces przyłączania grup alkilowych (metylowych) (-CH3) do zasad azotowych nukleotydów, w szczególności do cytozyny, rzadziej do adeniny (w pozycji 6). Produktem metylacji cytozyny jest zazwyczaj 5-metylocytozyna, czasem N4-metylocytozyna. W proces ten zaangażowane są enzymy z grupy metylaz DNA.

cytozyna
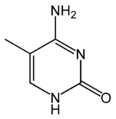
5-metylocytozyna

N4-metylocytozyna
Istnieje wiele technik służących do badania tego procesu. Przykładowo, można wykorzystać niektóre techniki immunocytochemiczne, ponieważ udało się opracować specyficzne przeciwciała zdolne do wiązania się z 5-metylocytozyną.
Metylacja jest odpowiedzialna za wiele procesów związanych z rozwojem organizmów – w szczególności za wytworzenie i utrzymywanie odpowiedniej specyfikacji tkankowej.
Wzrost metylacji części regulatorowej genu może wywołać zahamowanie jego ekspresji; ogólnie zakłada się, że wysoka metylacja pewnych fragmentów chromatyny związana jest z jej częściową lub całkowitą inaktywacją transkrypcyjną. Innymi słowy duży odsetek zmetylowanych nukleotydów stanowi sygnał dla białek uczestniczących w regulacji struktury chromatyny nakazujący inicjację procesów kondensacji chromatyny, co jest równoznaczne z przemianą euchromatyny w niedostępna dla czynników transkrypcyjnych heterochromatynę. W związku z powyższym metylacja chromatyny stanowi jeden z najważniejszych mechanizmów odpowiedzialnych za regulację ekspresji genów w przypadku organizmów jądrowych.
U ssaków około 5% reszt cytozynowych jest stale zmetylowanych, metylacja zachodzi zazwyczaj w dinukleotydowych sekwencjach CpG, często nazywanych "wyspami CpG". U roślin procent metylacji jest zazwyczaj większy – często wynosi on około 30%, proces ten może zachodzić w sekwencjach symetrycznych postaci wysp CpG i wysp CpNpG (gdzie N oznacza dowolny nukleotyd) oraz w sekwencjach niesymetrycznych w obrębie wysp CpNpN. Zazwyczaj metylacja u bezkręgowców obejmuje mniejszą część chromatyny, u niektórych organizmów takich jak Drosophila melanogaster nie zachodzi.
Przede wszystkim, poziom ekspresji danego genu jest skorelowany z ilością zmetylowanego DNA w sekwencji promotora. Im stopień metylacji jest większy tym słabsza ekspresja danego genu. Należy zauważyć, że geny mogą być aktywowane lub wygaszane przez dodawanie bądź usuwanie grup metylowych. Przykładowa silna metylacja transgenu przed wprowadzeniem go do komórki zazwyczaj prowadzi do zahamowania jego transkrypcji. Natomiast wprowadzenie do sekwencji DNA azacytozyny, będącej analogiem cytozyny, który nie może być zmetylowany w pozycji 5 prowadzi do aktywacji uprzednio zablokowanego genu. U ssaków i u roślin podczas wczesnych faz rozwoju genom przechodzi zmiany swego profilu metylacji. W przypadku ssaków wzór metylacji jest wymazywany w zygocie i ponownie generowany w epiblaście przed rozpoczęciem gastrulacji. U roślin zmiany w profilu metylacji są spowodowane przez transpozony (które z kolei są blokowane przez proces metylacji). Istnieją co najmniej dwa mechanizmy, poprzez które metylacja może prowadzić do blokowania ekspresji genów. W pierwszym dodanie grupy metylowej (która w oczywisty sposób zmienia przestrzenną strukturę rozpatrywanej sekwencji DNA) uniemożliwia przyłączenie się czynnika transkrypcyjnego, który w efekcie nie rozpoznaje swej docelowej sekwencji. W drugim mechanizmie zmetylowanie DNA prowadzi do przyłączenia specyficznych białek pokrywających DNA, co także uniemożliwia czynnikom transkrypcyjnym dostęp do chromatyny. Obecnie udało się wyizolować i zbadać właściwości wielu białek wiążących się do zmetylowanych sekwencji DNA.
Powszechnie uważa się, że metylacja DNA jest konsekwencją wyciszania transkrypcji, a nie czynnikiem inicjującym ten proces. Powstało kilka teorii tłumaczących rolę tego procesu; może ona blokować ogólne mechanizmy kontroli ekspresji genów, a także stanowić ochronę przed transkrypcją różnych form inwazyjnego DNA. Zarówno u roślin jak i u zwierząt transgeny często są poddawane metylacji de novo co powoduje utratę ich funkcji.
Zazwyczaj mamy do czynienia z tzw. metylacją symetryczną (sekwencje postaci CpG lub CpNpG), te same sekwencje są metylowane w obu niciach DNA, (jeżeli będą odczytywane w przeciwległych kierunkach). Takie rozwiązanie znacznie ułatwia przekazywanie niezmienionego wzorca metylacji komórkom potomnym. Należy zauważyć, że zachowanie niezmienionego profilu metylacji jest konieczne do utrzymania prawidłowego profilu ekspresji białek w danej komórce. Jednakże ogólne zmiany we wzorze metylacji, zachodzące podczas wczesnego rozwoju ssaków muszą być związane z aktywnością de novo odpowiednich metylaz. Wzór metylacji ustala się jedynie raz w określonej linii komórek i następnie jest on sukcesywnie przekazywany w czasie kolejnych rund replikacyjnych poprzez aktywność innych metylaz DNA, zdolnych do rozpoznawania hemimetylowanego DNA (to znaczy takiego, w którym jedynie jedna nić zawiera zmetylowane nukleotydy). Udało się wyizolować kilka takich enzymów.
Poprzez metylację DNA kontrolowane są istotne procesy związane z rozwojem organizmów, takie jak imprinting oraz inaktywacja chromosomu X. Zaburzenia metylacji DNA mogą doprowadzić do przemiany nowotworowej komórki (na przykład w wyniku wyłączenia ekspresji niektórych genów supresorowych nowotworów)[1]. Badanie wzorca metylacji możliwe jest m.in. poprzez zastosowanie zmodyfikowanej reakcji PCR. Ponieważ większość tkanek posiada specyficzny dla siebie wzór metylacji pojawiły się pomysły na zastosowanie tego rodzaju badań w diagnostyce nowotworów.
Deaminacja 5-metylocytozyny
Deaminacja niemodyfikowanej cytozyny prowadzi do uracylu, który jest rozpoznawany przez enzymy naprawcze DNA (→ Naprawa DNA); natomiast w wyniku deaminacji 5-metylocytozyny powstaje tymina, co prowadzić może do mutacji typu tranzycji.
Zarówno cytozyna, jak i 5-metylocytozyna ulegają deaminacji pod wpływem kwasu azotawego, natomiast wodorosiarczyny deaminują jedynie cytozynę. Pozwala to na analizę metylacji DNA za pomocą sekwencjonowania wodorosiarczynowego[2].
Bibliografia
- R.M. Twyman Krótkie wykłady. Biologia rozwoju Wyd. PWN Warszawa 2003.
- Piotr Węgleński (red) Genetyka molekularna, wydanie 2 poprawione Wyd. PWN Warszawa 2006
Przypisy
- ↑ Elias Daura-Oller i inni, Specific gene hypomethylation and cancer: New insights into coding region feature trends, „Bioinformation”, 3 (8), 2009, s. 340–343, DOI: 10.6026/97320630003340, PMCID: PMC2720671.
- ↑ Susan J. Clark i inni, High sensitivity mapping of methylated cytosines, „Nucleic Acids Research”, 22 (15), 1994, s. 2990-2997, DOI: 10.1093/nar/22.15.2990, PMID: 8065911, PMCID: PMC310266.c?
Media użyte na tej stronie


