Rak gruczołu krokowego
| carcinoma prostatae | |
.jpg) Gruczolakorak prostaty (obraz mikroskopowy, barwienie hematoksyliną i eozyną) | |
| ICD-10 | C61 |
|---|---|
Rak gruczołu krokowego, rak prostaty, rak stercza – pierwotny nowotwór złośliwy gruczołu krokowego pochodzenia nabłonkowego. Jest to jeden z najczęstszych nowotworów złośliwych u ludzi, pod względem zapadalności u mężczyzn ustępujący jedynie rakowi płuca. Najczęstszym podtypem histopatologicznym jest gruczolakorak zrazikowy. Do czynników ryzyka rozwoju choroby zalicza się wiek, wywiad rodzinny obciążający w kierunku tego nowotworu oraz przynależność rasową i etniczną. Wczesny rak stercza nie daje żadnych objawów, natomiast jeśli one występują, to są podobne do objawów łagodnego rozrostu tego gruczołu i obejmują bolesne oddawanie moczu, w tym uczucie pieczenia, wrażenie stałego parcia na mocz, trudności w rozpoczęciu mikcji, wąski strumień moczu, częste oddawanie go w nocy, bóle w kroczu i w podbrzuszu, niekiedy zatrzymanie moczu.
W związku z upowszechnieniem się oznaczenia stężenia swoistego antygenu sterczowego (PSA) większość przypadków choroby jest rozpoznawana w stadium bezobjawowym. Rozpoznanie jest oparte o badanie histopatologiczne materiału tkankowego pobranego podczas biopsji, którą wykonuje się z powodu nieprawidłowego stężenia PSA lub stwierdzenia nieprawidłowości w badaniu przez odbytnicę. Wybór metody leczenia zależy od stopnia zaawansowania choroby, ocenionej kategorii ryzyka choroby, obciążeń chorobami współistniejącymi, wieku chorego i oceny wpływu leczenia na jakość życia chorego. W miejscowo ograniczonym raku prostaty (czyli występującym tylko w obrębie tego gruczołu) postępowanie może polegać na aktywnym nadzorze (aktywnej obserwacji) jako jedynym środku, radykalnym wycięciu stercza (radykalnej prostatektomii) lub radykalnej radioterapii. Leczenie choroby zaawansowanej z przerzutami opiera się na hormonoterapii polegającej na chirurgicznym lub farmakologicznym zablokowaniu produkcji androgenów nazywanym ablacją androgenową. W celu uzyskania ablacji androgenowej podaje się agonisty lub antagonisty gonadoliberyny. Pojawienie się oporności na hormonoterapię jest nazywane opornością na kastrację; w jej leczeniu stosuje się docetaksel, abirateron, enzalutamid, sipuleucel-T lub radioaktywny 223Ra.
Epidemiologia
Rak gruczołu krokowego jest jednym z najczęstszych nowotworów złośliwych u ludzi. Jest drugim najczęściej rozpoznawanym nowotworem u mężczyzn i pod względem częstości ustępuje jedynie rakowi płuc[1][2][3]. Co roku rozpoznaje się 1 100 000 nowych przypadków tego nowotworu, co stanowi aż 15% rozpoznań nowotworu złośliwego u mężczyzn[1][4].
Zapadalność na raka gruczołu krokowego rośnie z wiekiem. Przed 50. rokiem życia występuje bardzo rzadko i w tej grupie wiekowej dotyczy jedynie 0,1% chorych[5]. 85% zachorowań jest stwierdzanych po 65. roku życia[5]. Około 85. roku życia ogólne ryzyko zachorowania wynosi w zależności od regionu świata 0,5–20%[5]. W badaniach sekcyjnych częstość występowania jest jeszcze większa, ponieważ te ujawniają utajoną część przypadków w bardzo wczesnym stadium, które ze względu na długi okres przedkliniczny nigdy nie uległyby manifestacji, ponieważ część mężczyzn wcześniej umiera z innych przyczyn niż rak stercza[6][7]. W badaniach sekcyjnych zapadalność na raka gruczołu krokowego u osób poniżej 30. roku życia wynosi około 5% i rośnie nieliniowo wraz z wiekiem, około 40–50. roku życia wynosząc 15%, a około 85. roku życia blisko 60%[7].
Zapadalność na raka gruczołu krokowego wykazuje znaczne zróżnicowanie geograficzne i etniczne, a odsetki zapadalności w poszczególnych regionach świata mogą się różnić nawet 25-krotnie[1][2][3][5]. Różnice w zapadalności w poszczególnych regionach świata częściowo są związane z rozpowszechnieniem badania PSA[2][8].
Większość zachorowań (70%) obserwuje się w krajach wysoko rozwiniętych[1][2][3]. Do regionów o najwyższej zapadalności należą Australia, Ameryka Północna, Europa Zachodnia i Północna, gdzie notuje się roczny współczynnik zapadalności odpowiednio 111,6, 97,2, 94,9 i 85,0 zachorowań na 100 000[1][9]. Do regionów o relatywnie wysokiej zapadalności należą również Karaiby, Ameryka Południowa i Południowa Afryka (odpowiednio 79,8, 60,1, 61,7 zachorowań na 100 000)[1]. Niską zapadalność obserwuje się w Azji Południowo-Centralnej i Wschodniej, która wynosi odpowiednio 4,5 i 10,5 przypadków na 100 000[1]. Ogólnie od lat 80. obserwuje się wzrost zapadalności na raka gruczołu krokowego, co również jest wiązane z wprowadzeniem i upowszechnieniem badania PSA[3][10].
Około 10% chorych na raka gruczołu krokowego umiera z powodu tej choroby, ale ogólnie rak stercza stanowi jedną z wiodących przyczyn zgonów z powodu choroby nowotworowej[11]. Rak gruczołu krokowego jest piątą przyczyną zgonów z powodu choroby nowotworowej na świecie[2][3]. Co roku z powodu tego nowotworu umiera ponad 300 000 mężczyzn, co stanowi 6,6% zgonów z powodu choroby nowotworowej[1]. Najwyższą umieralność obserwuje się w populacjach pochodzenia afrykańskiego, szczególnie w Karaibach i Afryce Subsaharyjskiej, a najniższą w Azji[1][2][3][9][10].
Objawy kliniczne

Około 75% przypadków tego nowotworu jest wykrywanych w stadium bezobjawowym ograniczonym do narządu[12]. Wczesny rak gruczołu krokowego zwykle nie daje żadnych objawów[13]. Jeśli nowotwór stercza powoduje objawy, to są one podobne do objawów związanych z łagodnym rozrostem gruczołu krokowego[14][15][16].
Nowotwór może powodować utrudnienie odpływu moczu z pęcherza moczowego, co objawia się objawami dyzurycznymi obejmującymi bolesne oddawanie moczu, uczucie pieczenia w cewce moczowej (stranguria), stałego parcia na mocz (pollakisuria), mogą również występować trudności w rozpoczęciu mikcji, uczucie niepełnego wypróżnienia, wąski strumień moczu, częste oddawanie moczu w nocy (nykturia), bóle w kroczu i w podbrzuszu[14][15]. Sporadycznie pojawia się krwinkomocz[15], możliwe jest zatrzymanie moczu[17]. Objawy ze strony układu moczowego są związane z budową anatomiczną i przechodzeniem cewki moczowej przez zmieniony gruczoł krokowy. Z kolei takie objawy jak trudność w uzyskaniu wzwodu lub bolesna ejakulacja są związane z uchodzeniem nasieniowodów w części sterczowej cewki moczowej w obrębie stercza[13]. W chorobie zaawansowanej z przerzutami występują bóle kostne, złamania patologiczne, zespół ucisku rdzenia kręgowego, niedokrwistość, utrata masy ciała[14][15][17].
W badaniu fizykalnym największe znaczenie ma badanie palcem przez odbytnicę. Może ono wykazać wyczuwalny guzek w gruczole krokowym, nadmierną twardość gruczołu i naciek nowotworowy obejmujący kości[17]. Typowo zmiany występują w tylnych i bocznych częściach stercza, manifestują się jako niesymetryczne zgrubienia lub guzki. Około 25–35% przypadków guzów gruczołu krokowego jest niebadalna palpacyjnie w badaniu per rectum[15]. Brak objawów w badaniu per rectum nie wyklucza nowotworu. U około 25–50% chorych z nieprawidłowościami w badaniu choruje na nowotwór gruczołu krokowego[18].
Czynniki ryzyka
Do dobrze poznanych czynników ryzyka rozwoju raka gruczołu krokowego zalicza się wiek, wywiad rodzinny w kierunku tego nowotworu oraz przynależność rasową i etniczną[19][20]. Ponadto badania epidemiologiczne sugerują kilka czynników środowiskowych i dietetycznych mogących mieć wpływ na ryzyko rozwoju tego nowotworu[21]. Do potencjalnych czynników ryzyka rozwoju tego nowotworu zalicza się dietę bogatą w mięso lub nabiał, otyłość, przebycie choroby przenoszonej drogą płciową, palenie tytoniu oraz ekspozycję zawodową na niektóre czynniki chemiczne[22][23][21][24][25].
Wiek
Wiek jest silnym czynnikiem ryzyka zachorowania, a także zgonu z powodu tej choroby[20]. Ryzyko zachorowania na nowotwór gruczołu krokowego rośnie wraz z wiekiem[10]. Do większości zachorowań dochodzi powyżej 65. roku życia, a przed 50. rokiem życia jest bardzo rzadki[5].
Rasa
Ryzyko zachorowania różni się w poszczególnych populacjach. Najwyższe ryzyko zachorowania jest stwierdzane u osób rasy czarnej, które jest wyższe niż u osób rasy białej[26]. Jest to szczególnie zauważalne u Afroamerykanów, u których stwierdza się 2,5-krotnie większe ryzyko zachorowania niż u osób białych[21]. U osób rasy czarnej rak stercza jest rozpoznawany na wyższym stopniu zaawansowania i w tej grupie chorych obserwuje się wyższą śmiertelność z powodu choroby[26]. Również wysokie ryzyko obserwuje się u osób rasy białej[20], najniższe odsetki zapadalności obserwuje się w populacjach azjatyckich[5].
Występowanie rodzinne
Występowanie raka gruczołu krokowego w rodzinie jest silnym czynnikiem ryzyka zachorowania na ten nowotwór[27][28][29]. Nawet u 20% chorych na raka gruczołu krokowego stwierdza się występowanie tego nowotworu u ojca lub braci chorego[30][31]. U krewnych chorych pierwszego stopnia, a więc u synów, braci i ojca chorego na raka gruczołu krokowego występuje 2–3-krotnie zwiększone ryzyko zachorowania[32][33]. Ryzyko zachorowania rośnie wraz z liczbą dotkniętych członków rodziny i niższym wiekiem zachorowania, ponadto ryzyko jest większe u braci chorego niż u syna chorego[29].
Dziedziczny rak gruczołu krokowego
Rodzinnym występowaniem raka gruczołu krokowego określa się akumulację przypadków tej choroby u blisko spokrewnionych członków określonej rodziny. Dziedziczny rak gruczołu krokowego (ang. hereditary prostate cancer) jest specyficzną grupą rodzinnego raka gruczołu krokowego, w którym stwierdza się określony wzór dziedziczenia genów podatności na chorobę[34][35][36]. Postać dziedziczna odzwierciedla obecność mutacji w wysokim stopniu predysponującym do raka, z kolei postać rodzinna poza mutacjami predysponującymi do choroby również odzwierciedla obecność czynników środowiskowych[36].
Ocenia się, że w około 5–10% przypadków raka gruczołu krokowego, a w populacji o wczesnym początku choroby nawet u 30–40% chorych, jest ona związana z genami o wysokiej predyspozycji dziedziczonymi autosomalnie dominująco. Ponadto wiadomo, że za postać dziedziczną odpowiadają rzadsze mutacje genowe dziedziczone autosomalnie recesywne o wysokiej penetracji lub dziedziczone sprzężone z chromosomem X[37][38][39][40].
Ze względu na trudność w określeniu odpowiedzialnych za zespół genów, dziedziczny rak gruczołu krokowego jest definiowany klinicznie za pomocą kryteriów rodowodowo-klinicznych[37][38]:
- rak gruczołu krokowego występuje u co najmniej 3 krewnych I stopnia,
- rak gruczołu krokowego występuje w co najmniej 3 kolejnych pokoleniach w rodzinie ojca lub matki,
- rak gruczołu krokowego występuje przynajmniej u 2 krewnych I lub II stopnia poniżej 55. roku życia.
Typowymi cechami dziedzicznego raka gruczołu krokowego jest młodszy wiek zachorowania w porównaniu do przypadków raka sporadycznego, średnio poniżej 55. roku życia, oraz wyższe ryzyko zgonu z powodu tej choroby[41]. Zwiększone ryzyko wystąpienia innych nowotworów w dziedzicznym raku gruczołu krokowego jest niejednoznaczne[41][38].
Czynniki dietetyczne
Niektóre badania sugerują, że wysokie spożycie produktów mlecznych wiąże się ze zwiększonym ryzykiem zachorowania[42][43]. Korelacja między wysokim spożyciem nabiału a ryzykiem raka gruczołu krokowego może być spowodowana wysokim spożyciem wapnia[19]. Za zwiększonym ryzykiem raka stercza u osób spożywających wysokie ilości wapnia przemawia wynik metaanalizy oraz wyniki innych badań[44][45], ale inne nie potwierdzają takiego związku[46][19].
Wyniki kilku badań kohortowych sugerują zwiększone ryzyko zachorowania u osób z wysokim spożyciem mięsa[47]. Jednak dwie metaanalizy kilkunastu badań nie potwierdzają związku wysokiego spożycia mięsa ze zwiększonym ryzykiem zachorowania na raka gruczołu krokowego[48][49][47][19]. Nie wykazano związku między spożyciem długołańcuchowych wielonienasyconych kwasów tłuszczowych omega-3 a zwiększonym ryzykiem zachorowania[50][51].
Otyłość
Otyłość zwiększa ryzyko rozwoju raka gruczołu krokowego[52]. Trzy metaanalizy wskazują na wzrost ryzyka względnego od 1,01 na każdy wzrost BMI 1 kg/m² do 1,05 na 1 kg/m²[53][54][55][52]. W badaniach amerykańskich, gdzie stosuje się badania przesiewowe oparte na ocenie stężenia PSA, nie obserwuje się wpływu otyłości na ryzyko raka gruczołu krokowego, co jednak może być związane z mniejszymi stężeniami PSA u osób otyłych i niższym prawdopodobieństwem wykonania biopsji u takich chorych i rozpoznania choroby niż u osób nieotyłych, co przekłada się na wyniki badań epidemiologicznych[52]. U chorych na raka gruczołu krokowego wykazano związek otyłości ze zwiększoną śmiertelnością swoistą dla nowotworu[56][52].
Choroby przenoszone drogą płciową
W licznych badaniach kliniczno-kontrolnych wykazano związek między rakiem gruczołu krokowego a chorobami przenoszonymi drogą płciową, szczególnie rzeżączką i kiłą[10][57], jednak większość z tych badań miała charakter retrospektywny[19]. Na zwiększone ryzyko raka u osób, które przebyły zakażenie rzeżączką wskazują wyniki kilku metaanaliz i badań prospektywnych[58][59][60]. Z kolei inne badania nie potwierdzają takiej zależności[61][62].
Aktywność seksualna
Kilka badań wskazuje na ochronny wpływ wysokiej aktywności seksualnej ocenianej jako ilość ejakulacji w miesiącu[63][64][65]. Jednak opublikowano również wyniki badań nie potwierdzające takiego efektu ochronnego, a wręcz sugerujące zwiększone ryzyko zachorowania[57].
Aktywność fizyczna
Wiele badań wskazuje na ochronny wpływ aktywności fizycznej na ryzyko rozwoju raka gruczołu krokowego, choć część badań nie potwierdza takiego efektu i wpływ aktywności fizycznej na ryzyko raka stercza nie jest ostatecznie ustalony[66][67][19].
Palenie tytoniu
Rola palenia tytoniu w ryzyku rozwoju raka stercza nie jest jasna[47][19][10][68]. Z tego względu wielu autorów nie wskazuje palenia tytoniu jako czynnika ryzyka raka prostaty[19][10]. W metaanalizie 24 badań u nałogowych palaczy stwierdzono zwiększone ryzyko rozwoju raka gruczołu krokowego[69]. W innej metaanalizie i przeglądzie systematycznym zaobserwowano odwrotną korelację między paleniem tytoniu a ryzykiem raka, natomiast wykazano zwiększoną śmiertelność palących tytoń chorych na ten nowotwór. Może być to związane z tym, że palenie tytoniu sprzyja bardziej agresywnym postaciom nowotworów i chroni przed ich indolentnymi (to znaczy powolnymi i zwykle bezbolesnymi) formami[70][68]. Ze względu na zwiększenie śmiertelności chorych na raka gruczołu krokowego i gorsze wyniki leczenia zaleca się zaprzestanie palenia tytoniu[68].
Czynniki hormonalne
Wpływ androgenów na ryzyko raka gruczołu krokowego był oceniany w wielu badaniach, tylko w jednym zaobserwowano zwiększone ryzyko rozwoju nowotworu gruczołu krokowego u mężczyzn z wysokim stężeniem testosteronu[71][5], podczas gdy większość danych, w tym wyniki metaanaliz, nie potwierdzają roli wysokiego stężenia testosteronu w zwiększaniu ryzyka tego nowotworu[72][73][5].
IGF-1 wpływa na kontrolę proliferacji, różnicowania i apoptozy wielu typów komórek, w tym komórek gruczołu krokowego. W kilku badaniach powiązano wysokie stężenie IGF-1 z podwyższonym ryzykiem raka gruczołu krokowego[74][75][76][77][23].
Narażenie zawodowe
Wiele badań sugeruje korelację między wykonywanym zawodem a zwiększonym ryzykiem zachorowania na raka gruczołu krokowego[10]. W przeglądzie Doolan i współpracowników, ze względu na retrospektywny charakter większości badań, ich ograniczenia metodologiczne oraz brak badań prospektywnych, nie powiązano aktywności zawodowej ze zwiększonym ryzykiem zachorowania na tę chorobę[78]. Jednak ze zwiększonym ryzykiem zachorowania powiązano ekspozycję na kadm, arsen, związki chromu, ołów, policykliczne węglowodory aromatyczne i polichlorowane bifenyle[78].
Potencjalne czynniki protekcyjne
Spożywanie warzyw i owoców jest czynnikiem ochronnym przed wieloma typami nowotworów. Kilka dużych badań nie potwierdziło ochronnego wpływu warzyw i owoców na ryzyko zachorowania na raka gruczołu krokowego[79][80][81][19], jednak wyniki niektórych badań sugerują, że wysokie spożycie pewnych podgrup warzyw i owoców, jak warzywa kapustne, czy pewnych ich składników, jak likopen, może zmniejszać ryzyko zachorowania na ten nowotwór[19].
Likopen jest związkiem zaliczanym do karotenów[19]. Metaanaliza kilku badań wskazuje na ochronny efekt wysokiego spożycia likopenu[82], z drugiej strony badania porównujące likopen z placebo nie potwierdzają tego ochronnego wpływu[83][51]. Warzywa kapustne są bogate w glukozynolany, które poprzez indukowanie enzymów biorących udział w metabolizmie karcynogenów, mogą wykazywać działanie ochronne[19]. Dane kliniczne dotyczące ochronnego wpływu tych warzyw są niespójne, część badań sugeruje taki efekt[81], a inne badania go nie potwierdzają[84]. Soja i produkty sojowe ze względu na zawartość izoflawonów również mogą wykazywać efekt ochronny[19][23]. Metaanaliza wskazuje, że soja zmniejsza ryzyko zachorowania na nowotwór gruczołu krokowego, jednak efekt ochronny był obserwowany wyłącznie w populacjach azjatyckich, podczas gdy u osób rasy białej nie wykazano działania protekcyjnego soi[85][19]. Nie wykazano ochronnego wpływu witaminy E i selenu[86][51].
Zapobieganie
Proponowane modyfikowalne czynniki ryzyka są potencjalnym celem działań profilaktycznych w ramach profilaktyki pierwotnej[87]. W profilaktyce wtórnej mogą być stosowane badania przesiewowe, które mają na celu zmniejszenie śmiertelności związanej z rakiem gruczołu krokowego poprzez wczesne wykrycie choroby. Jednak ze względu na niepewny wpływ na zmniejszenie śmiertelności z powodu raka gruczołu krokowego oraz obawy o częste nadmierne rozpoznawanie przypadków raka niepowodujących zagrożenia dla życia chorego oraz ich niepotrzebne leczenie nie zaleca się populacyjnych badań przesiewowych w kierunku raka gruczołu krokowego[88][89][90][14][91]. Profilaktyka trzeciorzędowa koncentruje się na powstrzymaniu progresji i nawrotu choroby[92].
Profilaktyka pierwotna
Nie ma ustalonej strategii skutecznej profilaktyki pierwotnej raka gruczołu krokowego[93][94]. Działania profilaktyki pierwotnej raka gruczołu krokowego ogniskują się na modyfikacji potencjalnych czynników ryzyka związanych ze stylem życia. Badania epidemiologiczne wskazują pewien wpływ diety, niskiej aktywności fizycznej czy nadwagi na ryzyko rozwoju raka gruczołu krokowego, jednak taka asocjacja między tymi czynnikami ryzyka a ryzykiem choroby najprawdopodobniej nie jest silna[95][96]. Część autorów zaleca unikanie nadwagi, palenia tytoniu oraz stosowanie regularnej aktywności fizycznej[97]. Proponuje się dietę bogatą w warzywa i owoce, z niską zawartością mięsa i tłuszczów zwierzęcych, a także unikanie nadmiernej ilości nabiału[93].
Chemoprewencja
Inna badaną strategią profilaktyki raka gruczołu krokowego jest chemoprewencja, która polega na zastosowaniu określonej substancji pochodzenia naturalnego lub syntetycznego w celu zapobiegnięcia lub zahamowania procesu karcynogenezy[92][98]. Oceniano dwa typy czynników chemoprewencyjnych: inhibitory 5α-reduktazy (finasteryd, dutasteryd) oraz suplementacje witaminy E i selenu[99].
Inhibitory 5α-reduktazy takie jak finasteryd blokują przemianę testosteronu do dihydrotestosteronu i są wykorzystywane w leczeniu łagodnego rozrostu gruczołu krokowego (przerostu gruczołu krokowego)[99][92]. W badaniu klinicznym PCPT na blisko 19 tysiącach mężczyzn oceniono skuteczność chemoprewencji za pomocą finasterydu. Badanie wykazało spadek ryzyka zachorowania na nowotwór gruczołu krokowego o niskim stopniu złośliwości (poniżej 6 punktów w skali Gleasona) o 25%, lecz jednocześnie zanotowano niewielki wzrost ryzyka zachorowania na nowotwór o wysokim stopniu złośliwości[100][92][98][94]. Z drugiej strony możliwe jest, że finasteryd poprawiał skuteczność wykrywania nowotworów o wysokiej złośliwości, co przyczyniło się do zwiększenia liczby ich rozpoznań w tym badaniu[101][102]. W opublikowanych długoterminowych wynikach badania PCPT, choć potwierdzono zmniejszenie ryzyka raka, to nie zaobserwowano różnicy przeżycia całkowitego między leczonymi finasterydem a otrzymującymi jedynie placebo[103][101].
Badania III fazy sugerowały korzystny wpływ α-tokoferolu (witaminy E) oraz selenu w redukcji ryzyka raka gruczołu krokowego[104][101]. Jednak w badaniu SELECT na 35500 mężczyznach nie wykazano korzyści ze stosowania suplementacji witaminy E lub selenu[86][101]. Późniejsza analiza wyników tego badania sugeruje wręcz szkodliwy wpływ wysokiego stężenia selenu na ryzyko wystąpienia nowotworu o wysokim stopniu złośliwości[105].
Badania przesiewowe
Badanie przesiewowe (skriningowe, skrining, screening) jest metodą profilaktyki wtórnej polegającą na badaniu zdrowych osób bez objawów choroby w celu wczesnego wykrycia choroby, wczesnego rozpoczęcia leczenia, a przede wszystkim poprawy rokowania chorych[91]. Badania przesiewowe mogą być wykonywane poprzez zbadanie całej populacji (badanie populacyjne) lub zagrożonej populacji w oparciu o zindywidualizowany schemat ustalony przez lekarza[106]. Podstawowym i najważniejszym celem badania przesiewowego jest zmniejszenie śmiertelności z powodu raka poprzez jego wczesne wykrycie, natomiast celem samym w sobie nie jest samo zwiększenie wykrywalności i liczby chorych poddanych leczeniu[107]. Choć badanie przesiewowe prowadzi do zwiększenia wykrywalności raka gruczołu krokowego, rozpoznawania i leczenia raka we wcześniejszym stadium, to ze względu na specyficzną historię, z częstym łagodnym i wieloletnim przebiegiem, rak gruczołu krokowego stosunkowo rzadko jest powodem zgonu i bardzo niewielu chorych może odnieść rzeczywistą korzyść z badania przesiewowego[108]. Badania przesiewowe niosą ze sobą nadmierną wykrywalność (overdiagnosis) przypadków nieistotnego klinicznie raka nie niosącego zagrożenia dla życia chorego w perspektywie wielu lat oraz wiążą się z nadmiernym leczeniem (overtreatment) nieistotnych klinicznie raków, co pogarsza jakość życia[106].
Ze względu na niepewny wpływ na zmniejszenie śmiertelności z powodu raka gruczołu krokowego oraz obawy o częste nadmierne rozpoznawanie przypadków raka niepowodujących zagrożenia dla życia chorego oraz ich niepotrzebne leczenie nie zaleca się populacyjnych badań przesiewowych w kierunku raka gruczołu krokowego[88][89][90][14][91]. Przy czym mężczyznom z długim przewidywanym czasem przeżycia w dobrym stanie sprawności, po rozważeniu ryzyka nadrozpoznawalności przypadków raka indolentnego, może być oferowana zindywidualizowana strategia wczesnego wykrywania dostosowana do ryzyka[109]. Według EAU badania przesiewowe mogą być oferowane mężczyznom w wieku powyżej 50 lat z podwyższonym ryzykiem zachorowania na raka gruczołu krokowego o przewidywanym czasie przeżycia powyżej 15 lat lub mężczyznom powyżej 45. roku życia z rodzinnym wywiadem raka gruczołu krokowego lub mężczyzn rasy afroamerykańskiej w wieku powyżej 45 lat. Ponadto badania mogą być oferowane u mężczyzn w wieku powyżej 40. roku życia ze stężeniem PSA powyżej 1 ng/ml lub mężczyznom w wieku powyżej 60. roku życia ze stężeniem PSA powyżej 2 ng/ml[109].
- Skuteczność badania przesiewowego
Najważniejszym celem badania przesiewowego jest zmniejszenie śmiertelności z powodu raka gruczołu krokowego oraz poprawa jakości życia[110][107]. Wiadomo, że odsetkowy spadek ogólnej śmiertelności z powodu raka gruczołu krokowego jest częściowo związany z szerokim stosowaniem badania stężenia PSA[111][110][89], jednak brakuje dowodów o wysokiej jakości (dowodów 1 stopnia), że badania przesiewowe są opłacalne kosztoefektywnie w zmniejszeniu śmiertelności z powodu raka gruczołu krokowego[110]. Dwa duże randomizowane badania oceniające skuteczność badania przesiewowego w kierunku raka gruczołu krokowego dały sprzeczne wyniki[112].
W europejskim badaniu ERSPC na 182 000 mężczyzn w wieku od 50 do 74 lat co 4 lata oceniano stężenie PSA i po 11 latach obserwacji w grupie poddanych badaniu przesiewowemu zaobserwowano 299 zgonów z powodu raka gruczołu krokowego oraz 462 zgony z tego powodu w grupie kontrolnej bez badania przesiewowego. Badanie przesiewowe przyczyniało się do zmniejszenia śmiertelności z powodu raka stercza, relatywne zmniejszenie ryzyka zgonu w grupie poddanych badaniu przesiewowemu wynosiło 21%, jednak aby zapobiec jednemu zgonowi z powodu raka stercza, konieczne było przebadanie 1055 mężczyzn oraz rozpoznanie 48 przypadków raka, aby zapobiec jednemu zgonowi z powodu choroby, co jest związane z łagodnym przebiegiem choroby[113][114][112][115]. W Göteborgu na 20 000 mężczyzn w wieku od 50 do 64 lat przeprowadzono początkowo niezależne badanie, a następnie znaczną część z tej grupy włączono do badania ERSPC, dlatego badania nie można traktować jako niezależne potwierdzające badanie od wyżej opisanego badania ERSPC[116]. Badanie przesiewowe polegało na badaniu stężenia PSA co dwa lata aż do 69 roku życia[112]. Po 14 latach obserwacji rak gruczołu krokowego został wykryty u 12,7% mężczyzn poddanych badaniu przesiewowemu i 8,2% mężczyzn w grupie kontrolnej. Badanie przesiewowe zmniejszało ryzyko zgonu z powodu tego nowotworu o 44%. Po osiemnastu latach badania oceniono, że 139 mężczyzn należy poddać badaniu przesiewowemu, aby zapobiec jednemu zgonowi z powodu raka stercza, oraz trzeba rozpoznać 13 przypadków raka, aby zapobiec jednemu zgonowi z powodu raka stercza[117][118][116]. Przyczynami lepszych wyników badań przesiewowych w Göteborgu niż ERSPC są niższy wiek badanych i mniejszy odsetek występowania niewyleczalnego raka gruczołu krokowego[116].
Amerykańskie badanie PLCO na 76 685 mężczyznach w wieku od 55 do 74 lat oceniło skuteczność badania przesiewowego polegającego na corocznej ocenie stężenia PSA przez 6 lat oraz corocznym badaniu per rectum przez 4 lata. Po 15 latach obserwacji wskaźnik wykrycia był nieznacznie wyższy w grupie mężczyzn poddanych badaniu przesiewowemu, jednak nie stwierdzono zmniejszenia śmiertelności z powodu raka gruczołu krokowego[119][120][121][122]. Ograniczeniem badania był wysoki odsetek osób poddanych ocenie stężenia PSA przed włączeniem do badania przesiewowego[14]. W dalszych analizach podgrup zmniejszenia śmiertelności z powodu raka doświadczali jedynie mężczyźni nieobciążeni lub obciążeni tylko jedną chorobą współistniejącą[123][122].
W przeglądzie systematycznym Cochrane stwierdzono, że badania przesiewowe w kierunku raka gruczołu krokowego sprzyjają wykrywaniu raka, także we wcześniejszym stadium, jednak po metaanalizie 5 randomizowanych badań klinicznych obejmujących 341 000 mężczyzn nie zaobserwowano poprawy przeżycia swoistego dla nowotworu[124][125][88]. Wpływ badań przesiewowych na jakość życia jest niejasny[126][127][128][88].
- Nadrozpoznawalność nieistotnego klinicznie raka
Niekorzystną konsekwencją badań przesiewowych opartych o ocenę PSA jest niepotrzebne rozpoznawanie przypadkowych, nieistotnych klinicznie raków gruczołu krokowego nie stwarzających istotnego zagrożenia życia chorego, które najprawdopodobniej nigdy nie ujawniłyby się jako kliniczna choroba dająca objawy, przerzuty, czy powodująca przedwczesny zgon chorego[129][130][131][132]. Z kolei nadmierna rozpoznawalność nieistotnego klinicznie raka stercza często prowadzi do zbędnego leczenia i pogorszenia jakości życia chorego[131][132].
PSA ma stosunkowo niewielką swoistość oraz niewielką dodatnią wartość predykcyjną, dlatego sama ścieżka diagnostyki przesiewowej opartej o PSA z wykonaniem biopsji systematycznej stercza w przypadku nieprawidłowego stężenia markera przyczynia się do wykrywania nieistotnych klinicznie nowotworów stercza[129]. W badaniu ERSPC przy stężeniu PSA poniżej 3 ng/ml jedynie 25% badanych miało raka stercza, a pozostałej części badanych mężczyzn (75%) wzrost markera był związany z przyczynami nienowotworowymi, ponadto 72% rozpoznanych raków w badaniu przesiewowym było nowotworami niskiego ryzyka[113][133][134]. Zatem mimo że małe powolnie rosnące nowotwory nie wydzielają PSA, mogą być przypadkowo wykrywane w konsekwencji wzrostu PSA z przyczyn nienowotworowych lub wieku badanego[129]. Ponadto częstość występowania nienowotworowych przyczyn wzrostu stężenia PSA rośnie wraz z wiekiem, co zwiększa ryzyko wykrycia nieistotnych klinicznie raków stercza[129].
Częściowo nadrozpoznawalność jest związana z bardzo częstym rozwojem wysoko zróżnicowanych raków stercza cechujących się bardzo powolnym i bezobjawowym przebiegiem, z których w zdecydowanej większości nigdy nie rozwinie się objawowa lub zagrażająca życiu choroba[129][134]. Ponadto wiele przypadków raków o wysokiej złośliwości (niskim zróżnicowaniu) również ma powolny przebieg, który przekłada się na wieloletni okres konieczny do rozwoju klinicznej choroby[129]. Mimo niewielkiego zagrożenia dla życia ze strony takich łagodnych nowotworów, szczególnie u mężczyzn w zaawansowanym wieku, znaczna ich część może być wykryta podczas badania przesiewowego[129][134]. Niepotrzebne rozpoznawanie raków o powolnym przebiegu jest szczególnie niekorzystne u mężczyzn w zaawansowanym wieku lub ze współistniejącymi chorobami ograniczającymi przeżycie, którzy ze względu na ograniczoną przewidywaną długość życia nie odnoszą korzyści z rozpoznawania i leczenia raków o łagodnym przebiegu[135]. Z kolei nadrozpoznawalność raków gruczołu krokowego często prowadzi do niepotrzebnego agresywnego leczenia, choć część chorych ze względu na wieloletni naturalny przebieg choroby oraz zaawansowany wiek nie odnosi korzyści z takiego leczenia, które pogarsza jakość życia[135].
- Wiek oraz odstępy między badaniami
Nie ustalono optymalnego wieku włączenia do badania przesiewowego ani wieku w którym należy zaprzestać tego badania[136][90]. Badania oceniające skuteczność badań przesiewowych skupiały się na mężczyznach w wieku od 55 do 69 lat, przy czym badania ERSPC i z Göteborga wskazywały na redukcję ryzyka zgonu w grupach odpowiednio od 55 do 69 lat oraz od 50 do 64 lat, co przemawia za badaniem przede wszystkim mężczyzn w wieku 50–55 lat[137]. Przeprowadzone badania dowodziły korzyści z badań przesiewowych do 70. roku życia[138]. EAU sugeruje wykonywanie badań przesiewowych jedynie u mężczyzn z przewidywanym czasem przeżycia powyżej 15 lat[109], z kolei NCCN sugeruje takie badanie jedynie u mężczyzn z przewidywanym czasem przeżycia powyżej 10 lat[138].
Również nie ustalono optymalnego schematu wykonywania kolejnych oznaczeń PSA, który dawałby największe korzyści ze zmniejszenia umieralności z powodu raka stercza oraz nie powodowałby nadrozpoznawania choroby. NCCN u mężczyzn ze stężeniem PSA poniżej 1 ng/ml proponuje wykonywanie kolejnych oznaczeń w odstępach 2–4 letnich, a u mężczyzn z PSA 1–3 ng/ml co 1–2 lata[139].
- Badanie przesiewowe oparte o grupy ryzyka
Wykorzystanie badania przesiewowego opartego o PSA w grupach o zwiększonym ryzyku zachorowania na raka stercza może poprawić skuteczność badania przesiewowego[140][141]. Do grupy o podwyższonym ryzyku zachorowania należy wywiad rodzinny występowania raka gruczołu krokowego oraz pochodzenie afroamerykańskie[142]. Nie ma opublikowanych wyników badań oceniających skuteczność badań przesiewowych w grupach o wysokim ryzyku zachorowania. W badaniu PLCO nie analizowano podgrup z obciążąjącym wywiadem rodzinnym lub osób rasy afroamerykańskiej, a z kolei w ERSPC nie raportowano o pochodzeniu rasowym i dodatnim wywiadzie rodzinnym[142]. Kolejną metodą są indywidualne kalkulatory ryzyka, które mogą pomóc zredukować ryzyko niepotrzebnej biopsji[109][140]. Jednak żaden z dostępnych algorytmów oceniających ryzyko nie wykazał przewagi nad samodzielnym badaniem PSA[143].
Profilaktyka trzeciorzędowa
W profilaktyce trzeciorzędowej zaleca się zaprzestanie palenia tytoniu, stosowanie diety bogatej w warzywa i owoce, ograniczenie spożycia mięsa i nabiału, unikanie siedzącego trybu życia i podejmowanie codziennej aktywności fizycznej[144].
Histopatologia
.jpg)
.jpg)

W około 90–95% przypadków raka gruczołu krokowego u ludzi rozpoznaje się gruczolakoraka zrazikowego[145][146][147]. Rak rozwija się najczęściej obwodowo w części tylnej i tylno-bocznej[147]. Strefa obwodowa jest lokalizacją 68% raków, strefa przejściowa 24%, a centralna 8%[145]. Zwykle (w 85% przypadków) rak jest wieloogniskowy, a jego poszczególne ogniska mogą wykazywać różnice w obrazie histopatologicznym[147][146].
Klasyfikacja WHO nowotworów gruczołu krokowego[148][149]:
- nowotwory pochodzenia nabłonkowego (epithelial tumours):
- nowotwory gruczołowe:
- rak gruczołowy (gruczolakorak) zrazikowy (acinar adenocarcinoma, ICD-O 8140/3),
- rak sarkomatoidny (carcinoma with spindle cell differentiation, carcinosarcoma, sarcomatoid carcinoma, 8572/3),
- rak gruczołowy (gruczolakorak) przewodowy (ductal adenocarcinoma, 8500/3),
- nowotwory urotelialne (urothelial tumours):
- rak urotelialny (urothelial carcinoma, 8120/3),
- nowotwory płaskonabłonkowe (squamous tumours):
- rak gruczołowopłaskonabłonkowy (adenosquamous carcinoma, 8560/3),
- rak płaskonabłonkowy (squamous cell carcinoma, 8070/3),
- nowotwory podstawnokomórkowe (basal cell tumours):
- gruczolak podstawnokomórkowy (basal cell adenoma, 8147/0),
- gruczolakorak podstawnokomórkowy (basal cell carcinoma, 8147/3),
- rak gruczołowy (gruczolakorak) zrazikowy (acinar adenocarcinoma, ICD-O 8140/3),
- nowotwory neuroendokrynne (neuroendocrine tumours):
- gruczolakorak z różnicowaniem neuroendokrynnym (endocrine differentiation within adenocarcinoma, 8574/3),
- rakowiak (carcinoid tumour, 8240/3),
- rak drobnokomórkowy (small cell carcinoma, 8041/3),
- przyzwojak (paraganglioma, 8680/1),
- nerwiak zarodkowy (neuroblastoma, 9500/3),
- nowotwory gruczołowe:
- nowotwory zrębu gruczołu krokowego (prostatic stromal tumours):
- nowotwór podścieliska o niepewnym potencjale złośliwości (stromal tumour of uncertain malignant potential, 8935/1),
- mięsak podścieliska (stromal sarcoma, 8935/3),
- nowotwory mezenchymalne,
- chłoniaki i białaczki,
- różne nowotwory gruczołu krokowego.
Rak gruczołowy zrazikowy


Większość guzów powstaje w strefie obwodowej gruczołu krokowego[150][151]. W 85% przypadków nowotwór jest wieloogniskowy[152]. Makroskopowo guzy są trudne do odróżnienia od otaczającej tkanki, twarde, lite, na przekroju barwy od białoszarej do żółtej[153][151]. Guzy mogą deformować obrys narządu. Typowo guzy widoczne makroskopowo mają tendencję do wyższego stopnia zaawansowania i złośliwości w porównaniu do zmian niewidocznych makroskopowo, które często klinicznie nie były badalne palpacyjne. W obrazie makroskopowym rzadko są obecne obszary krwotoczne lub martwicy[153]. Typowo nowotwór szerzy się poza granicę zasięgu makroskopowego[153].
W obrazie mikroskopowym gruczolakoraki gruczołu krokowego znacznie różnią się stopniem zróżnicowania (złośliwości histologicznej), dając spektrum nowotworów od dobrze zróżnicowanych (o niskiej złośliwości), które trudno odróżnić od prawidłowego utkania gruczołu krokowego do zmian o niskim zróżnicowaniu (o wysokiej złośliwości)[154]. Cechą wspólną dla wszystkich raków stercza jest obecność pojedynczej warstwy jednego typu komórek bez warstwy podstawnej, która jest widoczna w prawidłowych cewkach gruczołowych[155][151].
W dobrze zróżnicowanych nowotworach komórki nowotworowe tworzą stłoczone cewki gruczołowe o nieregularnym kształcie i różnej wielkości, które są wyścielone przez jedną warstwę komórek. Cewki gruczołowe są ułożone w dość bezładny sposób[155]. Kolejnym wzorem utkania typowym dla nacieku jest obecność atypowych struktur gruczołowych między dużymi, prawidłowymi gruczołami. Wraz z utratą różnicowania i tworzeniem struktur sitowatych, połączonych gruczołów lub słabo uformowanych gruczołów, coraz bardziej zaznacza się różnica między łagodnymi gruczołami a strukturami tworzonymi przez komórki nowotworowe[155]. Nisko zróżnicowane nowotwory są zbudowane z litych arkuszy lub sznurów komórek, a cewki gruczołowe są nieliczne lub poronne[155][156].
Komórki cechują się obecnością powiększonego jądra komórkowego i powiększonego jąderka. W niektórych komórkach nie jest obecne powiększone jąderko, jednak pozostają powiększone i hiperchromatyczne[157][158]. Figury mitotyczne mogą być liczne w nowotworach o wysokiej złośliwości[157]. Cytoplazma komórek budujących struktury gruczołowe jest amfifilna, w jej obrębie nie stwierdza się lipofuscyny[157].
Może być widoczna inwazja naczyń krwionośnych i chłonnych oraz nerwów, która jednoznacznie przemawia za złośliwym charakterem nowotworu[158]. Innymi cechami wskazującym na złośliwość nowotworu jest obecność struktur przypominających kłębuszki oraz niewielkich guzków zbudowanych z eozynofilnych włókien w obrębie cewek gruczołowych (mucinous fibroplasia, collagenous micronodules)[159][158]. W cewkach gruczołowych bywa obecna mucyna, ale jej obecność nie jest swoista dla raka. W guzach o niskiej złośliwości są obecne krystaloidy (prostatic crystalloids) widoczne jako ostre, gęste eozynofilne struktury o różnych kształtach geometrycznych obecne w świetle cewek, jednak ich obecność nie jest charakterystyczna dla raka[157][158].
Rak gruczołowy przewodowy

Jest to podtyp gruczolakoraka zbudowany z dużych gruczołów wyłożonych wysokimi komórkami walcowatymi. W postaci czystej gruczolakorak przewodowy stanowi jedynie 0,2–0,8% raków gruczołu krokowego u ludzi, ale znacznie częściej jest obecny jako komponent gruczolakoraka zrazikowego[160]. Gruczolakorak przewodowy może występować w centralnej części stercza lub częściej w jej obwodowej części, współwystępując z domieszką gruczolakoraka zrazikowego[160].
Makroskopowo guzy położone w centralnej części stercza są widoczne jako egzofityczna, polipowata albo brodawkowata zmiana wnikająca do cewki moczowej w okolicy wzgórka nasiennego. Guzy położone w obwodowej części są podobne do gruczolakoraka zrazikowego[161].
W obrazie mikroskopowym utkanie nowotworu budują wysokie komórki walcowate z obfitą najczęściej amfofilową cytoplazmą, które tworzą warstwy komórek przypominające utkanie endometrium. Czasem liczba mitoz jest wysoka i występuje znaczna atypia cytologiczna, w innych przypadkach liczba mitoz jest skąpa, a atypia minimalnie zaznaczona[161]. Nowotwór wykazuje różnorodność wzorów architektonicznych, które mogą być wymieszane. Wzór brodawkowaty najczęściej występuje w guzach położonych w centralnej części stercza, ale może być obecny także w guzach pojawiających się w jego obwodowej części. Wzór sitowaty jest utworzony przez duże przylegające do siebie gruczoły, które tworzą szczelinowate otwory. Wzór lity jest powiązany z występowaniem innych wzorów architektonicznych gruczolakoraka przewodowego, cechuje się występowaniem litych gniazd komórek nowotworowych pooddzielanych niepełnymi przegrodami lub rdzeniami włóknisto-naczyniowymi[162].
Rak urotelialny gruczołu krokowego

Jest to nowotwór pochodzący z nabłonka dróg moczowych w obrębie gruczołu krokowego. Pierwotny rak urotelialny obejmujący stercz stanowi 0,7–2,8% nowotworów tego narządu u ludzi[163]. Zwykle jest związany z pierwotnym rakiem urotelialnym pęcherza moczowego lub urotelialnym rakiem moczowodu. Zajęcie gruczołu krokowego może być związane z szerzeniem się raka urotelialnego do stercza, implantacją nowotworu pochodzącego z pęcherza moczowego lub moczowodu albo następstwem niezależnej wieloogniskowej karcynogenezy raka urotelialnego[164]. Rak urotelialny w obrębie stercza bez obecności raka urotelialnego w obrębie pęcherza moczowego jest rzadki[164].
Większość raków urotelialnych w obrębie stercza jest wysokiej złośliwości i są one związane z rakiem in situ[163]. Typowa jest obecność pagetoidalnego szerzenia się pojedynczych komórek lub zagnieżdżanie się komórek nowotworowych między warstwą podstawną i warstwą komórek wydzielniczych[163]. Przy rozległym naciekaniu rak urotelialny może wypełniać i poszerzać kanaliki gruczołu krokowego[163][164]. Może być obecna martwica typu comedo[164]. Komórki nowotworowe charakteryzują się wyraźnym pleomorfizmem jądrowym, hiperchromazją i obecnością licznych mitoz. W przypadku inwazji podścieliska obecne są nieregularne gniazda lub sznury komórek i wyraźna reakcja desmoplastyczna[163][164].
Rak płaskonabłonkowy i gruczołowopłaskonabłonkowy
Są to nowotwory wykazujące cechy różnicowania w kierunku nabłonka płaskiego. Rak płaskonabłonkowy i gruczołowopłaskonabłonkowy stanowią mniej niż 0,6% nowotworów stercza u ludzi. Najczęściej występują w strefie przejściowej gruczołu krokowego[165]. Z definicji rak płaskonabłonkowy nie może zawierać komponentu gruczołowego i musi wykazywać obecność cechy keratynizacji oraz obecność mostków międzykomórkowych[166].
Makroskopowo zwykle są to stosunkowo duże guzy, które mogą zastępować znaczną część miąższu stercza. Guzy są lite, twarde, na przekroju koloru białożółtego lub szarobrązowego[164]. W obrazie mikroskopowym czystego raka płaskonabłonkowego występują gniazda, pasma lub arkusze wielobocznych komórek z wyraźną atypią jądrową z widocznymi cechami różnicowania w kierunku raka płaskonabłonkowego obejmującymi keratynizację, tworzenie pereł keratynowych lub mostków międzykomórkowych. W obrazie raka gruczołowopłaskonabłonkowego mogą być obecne wyraźnie odrębne komponenty płaskonabłonkowy i gruczołowy lub utkanie może bezpośrednio w siebie przechodzić[164].
Rak podstawnokomórkowy
Jest to nowotwór zbudowany z komórek podstawnych[167]. Rak podstawnokomórkowy gruczołu krokowego jest rzadkim nowotworem, dotyczy przede wszystkim starszych mężczyzn[164][167]. Makroskopowo nowotwór jest twardy i lity[164]. W obrazie mikroskopowym spotyka się kilka odmiennych wzorów utkania[164]. Część guzów reprezentuje utkanie podobne do raka podstawnokomórkowego skóry z obecnymi dużymi gniazdami komórek podstawnych z obwodowym palisadowaniem i obecną martwicą, z kolei inne wzory przypominają hiperplazję[164][167].
Nowotwory neuroendokrynne gruczołu krokowego
Nowotwory neuroendokrynne gruczołu krokowego obejmują raka drobnokomórkowego oraz rakowiaka. Ponadto wyróżnia się ogniskowe różnicowanie raka w kierunku nowotworu neuroendokrynnego. Nowotwory neuroendokrynne gruczołu krokowego wykazują podobne objawy kliniczne jak inne nowotwory stercza, choć możliwe jest występowanie zespołów paraneoplastycznych, jak zespół Cushinga, zespół nieprawidłowego wydzielania hormonu antydiuretycznego, zespół Lamberta-Eatona, hiperkalcemia[168].
- Gruczolakorak z różnicowaniem neuroendokrynnym
W wielu przypadkach gruczolakoraków gruczołu krokowego obecne są cechy różnicowania pojedynczych komórek w kierunku nowotworu neuroendokrynnego. W 5–10% przypadków gruczolakoraka występują obszary z licznymi skupiskami lub pojedynczymi komórkami neuroendokrynnymi[169][168].
- Rak drobnokomórkowy
Większość nowotworów nie wykazuje czynności hormonalnej, ale część wydziela ACTH lub wazopresynę. Histologicznie nowotwór jest podobny do raka drobnokomórkowego płuca. W połowie przypadków występuje mieszane utkanie raka drobnokomórkowego i gruczolakoraka[170].
- Rakowiak
Rakowiak gruczołu krokowego jest wyjątkowo rzadkim nowotworem, nowotwór wykazuje typowe cechy morfologiczne charakterystyczne dla rakowiaków[171].
Skala Gleasona




Opracowano wiele systemów oceny złośliwości histologicznej nowotworów gruczołu krokowego, które opierają się o architekturę zmian, a część z nich uwzględnia również stopień atypii[172]. Zalecanym systemem oceny złośliwości raka stercza jest skala Gleasona[173][172]. Oparta jest ona na stopniu architektonicznego zróżnicowania raka zależny od obrazu cewek gruczołowych, granicy guza z otoczeniem i naciekania podścieliska[174][172]. Wyróżniono 5 stopni, w których stopień 1 oznacza bardzo dobrze zróżnicowanego raka, a 5 stopień raka nisko zróżnicowanego[175]. Ze względu na heterogenne utkania nowotworu ocenie podlega wzorzec pierwotny i wtórny, czyli najbardziej dominujący wzorzec histoarchitektoniczny i drugi najczęstszy wzorzec. Wynik przedstawia się za pomocą sumy punktów za stopnie wzorca pierwotnego i wtórnego. Jeśli nowotwór posiada wyłącznie jedno utkanie to wynik podaje się przez podwojenie wyniku pojedynczego wzoru. Wynik 2 i 3 jest rzadki, co jest związane z rzadkim występowaniem typu 1, wynik 4 również jest względnie rzadki. Najczęściej rozpoznaje się nowotwory w stopniach 6 i 7[172]. Niskiemu stopniowi złośliwości histologicznej odpowiadają wyniki ≤ 6, a wysokiemu stopniowi złośliwości wyniki ≥ 7[147].
- Stopień 1
W stopniu 1 widoczne są dobrze odgraniczone guzki zbudowane ze ściśle upakowanych jednolitych struktur gruczołowych o równej wielkości, okrągłego lub owalnego jednolitego kształtu. Nowotworowe cewki gruczołowe nie naciekają sąsiedniej łagodnej tkanki gruczołowej. Wzór stopnia 1 jest dość rzadki[176][177][173].
- Stopień 2
W stopniu 2 utkanie budują okrągłe lub owalne gładko zakończone struktury gruczołowe. W porównaniu ze stopniem 1 gruczoły są mniej jednorodne i bardziej luźno ułożone[176]. Gruczoły mają pośrednią wielkość, obserwuje się pewną zmienność wielkości i kształtu gruczołów, przy czym jednak są większe niż w stopniu 3, a zmienność wielkości i odstępów między gruczołami mniejsza niż w stopniu 3[176][173]. Ogniska są dość ograniczone, jednak może występować minimalny naciek sąsiednich tkanek stercza[176][177]. Komórki w stopniu 1 i 2 zawierają obfitą ilość bladej cytoplazmy (atypia komórek nie wchodzi w zakres oceny według klasyfikacji Gleasona). Wzór stopnia 2 zwykle jest obecny w nowotworach w strefie przejściowej i tylko okazjonalnie w obrębie strefy obwodowej[176].
- Stopień 3
Jest to najczęstszy wzór histoarchitektoniczny. W porównaniu do wzoru 1 i 2 gruczoły nowotworowe wykazują wyraźne różnice wielkości i kształtu oraz odległości między nimi[176][173][177]. Typowo gruczoły są małe, ale mogą być obecne duże i nieregularne. Każdy gruczoł jest otoczony przez zrąb i ma otwarte światło[176]. Ułożenie gruczołów w zrębie jest losowe[173]. Nowotworowe gruczoły często wnikają między te prawidłowe[176].
- Stopień 4
W stopniu 4 gruczoły nowotworowe ulegają łączeniu, mogą tworzyć struktury sitowate[176]. Słabo zróżnicowane gruczoły mogą być pozbawione światła, bywają obecne struktury przypominające kłębuszki[176][177]. Połączone gruczoły nie są całkowicie oddzielone przez zrąb[176].
- Stopień 5
W stopniu 5 obserwuje się niemal całkowitą utratę różnicowania gruczołów[177]. Występuje niemal całkowita utrata światła gruczołów, które występuje jedynie sporadycznie. Komórki układają się w lite arkusze, pasma lub występują pojedynczo jako naciek zrębu. Może występować martwica typu centralnego (martwica typu comedo, comedonecrosis)[176].
Historia naturalna


Rak gruczołu krokowego nie jest chorobą jednorodną i wykazuje dość zróżnicowaną historię naturalną[178]. U znacznej części chorych rak gruczołu krokowego ma stosunkowo łagodny przebieg, choć po wieloletnim okresie choroby może dojść do lokalnej progresji choroby lub rozwoju przerzutów odległych[178][179].
Choroba cechuje się wieloletnim okresem przedklinicznym[180]. Najczęściej rak jest rozpoznawany jako mała zmiana, o niskim stopniu złośliwości, która cechuje się kilkuletnim okresem podwojenia jej wielkości[181]. Jednak u poszczególnych chorych przebieg choroby może być odmienny i u części z nich szybko dochodzi do progresji nowotworu[182][183].
W ponad 75% przypadków rak gruczołu krokowego jest wykrywany w stadium ograniczonym do narządu[12], w 85% przypadków jest on wieloogniskowy[146]. Większość raków jest zlokalizowana w części obwodowej stercza w pobliżu torebki gruczołu krokowego. Stosunkowo wcześnie dochodzi do inwazji torebki gruczołu krokowego i jej naciek jest stwierdzany w 80% przypadków raka[146].
Rak może szerzyć się przez ciągłość oraz za pośrednictwem naczyń krwionośnych lub limfatycznych[175]. Lokalne szerzenie się przez ciągłość najczęściej stwierdza się w części tylno-bocznej (guzy położone w obwodowej części gruczołu krokowego) oraz części przedniej (guzy strefy przejściowej)[153]. Nowotwory w obwodowej części gruczołu krokowego naciekają tkankę okołosterczową poprzez bezpośredni naciek torebki gruczołu krokowego lub szerząc się wzdłuż nerwów[153]. W dużych guzach może dojść do nacieku szyi pęcherza moczowego, co może doprowadzić do niedrożności szyi pęcherza[153]. Naciek pęcherzyków nasiennych może być następstwem bezpośredniego nacieku z zajętych tkanek miękkich lub szerzenia się wzdłuż przewodu wytryskowego[153]. Przegroda odbytniczo-pęcherzowa (powięź Denonvilliersa) stanowi barierę dla szerzenia się raka gruczołu krokowego i nowotwór rzadko się szerzy w kierunku tylnym[153], w przypadku spenetrowania tej przegrody nowotwór może naciekać odbytnicę[146].
Znacznemu zaawansowaniu miejscowemu zwykle towarzyszą przerzuty odległe[184]. Rak gruczołu krokowego najczęściej daje przerzuty do węzłów chłonnych, kości i płuc[185]. Szerzenie się drogą naczyń chłonnych skutkuje zajęciem węzłów chłonnych[185]. Zajęcie poszczególnych grup węzłów chłonnych odbywa się zgodnie z drenażem limfatycznym narządu[186]. Przerzuty w węzłach chłonnych najpierw stwierdza się w grupach węzłów poniżej rozwidlenia tętnicy biodrowej wspólnej: w węzłach biodrowych wewnętrznych i biodrowych zewnętrznych, następnie dochodzi do zajęcia węzłów biodrowych wspólnych, okołoaortalnych i pachwinowych[185]. Rozsiew drogą krwionośną skutkuje powstaniem przerzutów odległych, które najczęściej pojawiają się w szkielecie osiowym, a rzadziej w płucach, wątrobie i innych narządach[146]. Przerzuty w kościach zwykle mają charakter osteoblastyczny (80%), ale możliwe są zmiany osteoblastyczno-osteolityczne (15%) i osteolityczne (5%)[186][184]. Przerzuty kostne często lokalizują się w kręgosłupie, szczególnie w odcinku lędźwiowym i piersiowym, nasadach kości udowych, miednicy, żebrach, mostku i czaszce[184][185][153][186]. Przerzuty do płuc, wątroby i innych narządów występują raczej w późnym przebiegu choroby i dotyczą one odpowiednio 25% i 20% chorych[185][186].
Karcynogeneza
Karcynogeneza raka gruczołu krokowego, podobnie jak karcynogeneza innych nowotworów, jest wieloetapowym procesem związanym z nabywaniem i gromadzeniem kolejnych zmian cytogenetycznych w komórce nowotworowej prowadzących do zaburzenia funkcji niektórych genów[187][188]. Nabyte kolejne zmiany genetyczne w nieprawidłowej komórce sprzyjają zwiększeniu jej zdolności do podziału (proliferacji), zablokowaniu procesu różnicowania, oporności na apoptozę, nabyciu zdolności inwazyjnych i wytworzeniu przerzutów odległych[189]. Proces karcynogenezy jest wieloletni, pierwsze widoczne zmiany histologiczne bywają obecne już około 20. roku życia, podczas gdy klinicznie wykrywalny nowotwór pojawia się 3–4 dekady później[189].
W wyniku działania czynników uszkadzających, jak proces zapalny lub karcynogeny, dochodzi do śmierci komórki lub jej uszkodzenia i wyzwolenia procesów naprawczych. Sprzyja to pojawieniu się zmian genetycznych prowadzących do transformacji nowotworowej[188]. Podobnie jak w karcynogenezie innych procesów nowotworowych karcynogenezę raka gruczołu krokowego można podzielić na trzy etapy: inicjację, promocję i progresję. Etap inicjacji cechuje się obecnością uszkodzenia materiału genetycznego komórki, które może pozostać naprawione albo spowodować trwałą jego zmianę. Promocję charakteryzuje pojawianie się kolejnych mutacji i stymulacji proliferacji nieprawidłowej komórki. W etapie progresji dochodzi do gromadzenia licznych kolejnych zmian genetycznych, które wiążą się z inwazyjnością, stymulacją proliferacji i angiogenezy, zdolnością do wytwarzania przerzutów i utratę zależności od androgenów[190]. Najważniejszymi genami związanymi z karcynogenezą raka gruczołu krokowego są AR, CDKN1B, EZH2, KLK3, GSTP1, MYC, NKX3.1, TP53 i PTEN[191].
Podtypy genetyczne raka gruczołu krokowego
Rak gruczołu krokowego cechuje się brakiem wyróżniających podtypów histologicznych, podtypów różniących się rokowaniem czy odpowiedzią na leczenie. Najczęstszym podtypem jest gruczolakorak zrazikowy, w którym stwierdza się ekspresję AR, a pozostałe podtypy są rzadkie[192].
Jednak badania genetyczne pozwoliły wyróżnić kilka podtypów molekularnych raka gruczołu krokowego[193]. Nowotwory gruczołu krokowego są dzielone na związane ze zmianami w zakresie ETS (ERG, ETV1, ETV4, i FLI1) oraz związane z mutacjami SPOP, FOXA1 lub IDH1[194][195]. W nowotworach związanych z ETS często stwierdza zaburzenia szlaku PI3K/Akt, mutacje p53 oraz PTEN[196][195]. W nowotworach z mutacjami SPOP obserwuje się również zmiany w genie CHD1 i utratę chromosomu 2q i 6q[195].
Zmiany cytogenetyczne związane z rakiem gruczołu krokowego
- GSTP1
Gen koduje enzym transferazę glutationową, która bierze udział w metabolizmie wielu ksenobiotyków. Hipermetylacja promotora genu jest wczesnym zdarzeniem w karcynogenezie raka stercza i jest ona obserwowana w 70% przypadków śródnabłonkowej neoplazji stercza (PIN) oraz 90–95% przypadków raka gruczołu krokowego. Wyłączenie genu może skutkować zwiększoną ekspozycją na uszkodzenie materiału genetycznego i sprzyjać akumulacji kolejnych zmian cytogenetycznych[187].
- NKX3.1
Utrata heterozygotyczności chromosomu 8p21-22 zawierającego gen homeoboksowy NKX3.1 również jest wczesnym zdarzeniem w karcynogenezie raka gruczołu krokowego[187]. Pełni on rolę genu supresorowego[191]. Zmiany w obrębie tego genu dotyczą większości przypadków raka gruczołu krokowego[191]. Ocenia się, że dotyczy ona 15% przypadków śródnabłonkowej neoplazji stercza i nawet 85% przypadków raka gruczołu krokowego[187].
Gen pełni istotną rolę w procesie różnicowania komórek nabłonka gruczołu krokowego, a jego niedobór może się przyczyniać do zaburzenia różnicowania komórek, co może prowadzić do transformacji nowotworowej[187]. Całkowitą utratę ekspresji obserwuje się w przypadku progresji do raka, we wcześniejszym stadium jego obniżona ekspresja jest związana z mechanizmem utraty allelu lub metylacji promotora genu[197].
- ETS
Rodzina czynników transkrypcyjnych ETS wpływa na wiele procesów komórkowych, w tym proliferację, różnicowanie, apoptozę i angiogenezę[198]. Translokacja obejmująca gen kodujący czynnik transkrypcyjny ETS jest jedną z najczęstszych mutacji w raku gruczołu krokowego u ludzi[199]. Ocenia się, że występuje w 20% przypadków PIN i 50% przypadków gruczolakoraka[187][200]. W wyniku translokacji genu czynnika transkrypcyjnego z rodziny ETS i połączenia z zależnym od receptora androgenowego promotorem TMPRSS2 powstaje gen fuzyjny, który ulega nadekspresji i dereguluje wiele szlaków istotnych w karcynogenezie. W 90% rearanżacja ETS obejmuje gen ERG, rzadziej dochodzi do translokacji genu ETV1, ETV5 czy ETV4. Gen ERG zwykle ulega połączeniu z genem TMPRSS2, choć zidentyfikowano również inne geny regulowane androgenami, które ulegają fuzji z genem ERG[199]. Rola rearanżacji ETS w raku gruczołu krokowego jest niejasna. Sama nadekspresja ETS jest niewystarczająca do powstania raka i do powstania nowotworu konieczna jest akumulacja kolejnych mutacji[198][187].
- Receptor androgenowy (AR)

Aktywność receptora androgenowego (AR) jest ściśle związana z rakiem gruczołu krokowego[201]. Gen receptora jest zlokalizowany w obrębie Xq11-12. Receptor po związaniu z ligandem jakimi są testosteron lub dihidrotestosteron ulega zmianom konformacyjnym i dimeryzacji, a po przemieszczeniu do jądra komórkowego wpływa na regulację transkrypcji wielu genów odpowiadających za proliferację, apoptozę, różnicowanie i zdolność do inwazji[202][203][204]. Androgeny za pośrednictwem receptora androgenowego promują wzrost i proliferację komórek nowotworowych raka gruczołu krokowego[205]. Jednak nie stanowią wystarczającego czynnika do samodzielnej inicjacji karcynogenezy, choć są bardzo ważne w etapie progresji procesu karcynogenezy[206].
- Oporność na kastrację
Komórki nowotworowe raka gruczołu krokowego wymagają obecności androgenów do wzrostu i przetrwania, co jest podstawą leczenia opartego o deprywację androgenów, które zwykle pozwala uzyskać długotrwałą remisję choroby. Jednak rozwój oporności na deprywację androgenów i powstanie raka opornego na kastrację jest nieunikniony. Wyróżnia się cztery główne mechanizmy oporności na kastrację: zwiększona wrażliwość receptora androgenowego na jego agonistów, mutacje umożliwiające reakcję receptora androgenowego na alternatywne agonisty, zmiany powodujące aktywację receptora niezależnie od obecności agonisty oraz mechanizmy niezależne od receptora androgenowego[201].
Jednym z podstawowych mechanizmów oporności na deprywację androgenów jest wzrost ekspresji receptora androgenowego związany z amplifikacją jego genu[201]. Ocenia się, że amplifikacja genu kodującego receptor androgenowy może stanowić mechanizm oporności na kastrację blisko 30% przypadków takich nowotworów[207][201]. Powoduje to zwiększoną czułość receptora na niskie poziomy androgenów, które mimo właściwego leczenia deprywacyjnego są wystarczająco wysokie, aby podtrzymać aktywność receptora i zależne od niego szlaki[201]. Androgeny powstają in situ w obrębie guza oraz w ramach resztkowej funkcji nadnerczy. Ponadto obserwuje się obniżoną aktywność enzymów dezaktywujących androgeny[208][209][201].
Również kolejny mechanizm oporności jest związany z mutacją genu receptora androgenowego. W ich wyniku dochodzi do zmniejszenia selektywności ligandów aktywujących receptor, co powoduje jego aktywację pod wpływem innych hormonów steroidowych takich jak glikokortykoidy, estrogeny czy progesteron[201]. Kolejnym mechanizmem jest aktywacja receptora androgenowego przez mechanizmy niezależne od obecności ligandu. Ligandy receptora kinazy tyrozynowej takie jak IGF-1, KGF, EGF mogą aktywować receptor androgenowy poprzez aktywację szlaku PI3K/AKT/mTOR[201]. Nabycie oporności na kastrację może być związane z mechanizmami niezależnymi od receptora androgenowego. Czynniki prozapalne wydzielane przez komórki nowotworowe powodują naciek limfocytów, które z kolei wydzielają czynniki wpływające na aktywację szlaku STAT3 w komórkach nowotworowych. Aktywacja STAT3 promuje przeżycie komórek nowotworowych mimo braku aktywacji receptora androgenowego. Również regulacja w górę Bcl-2 chroni komórki nowotworowe przez apoptozą[201].
- PTEN i szlak PI3K/Akt/mTOR
Szlak PI3K/Akt/mTOR, regulując wzrost, przetrwanie i różnicowanie komórek, pełni kluczową rolę w karcynogenezie raka gruczołu krokowego. Ocenia się, że zaburzenia w obrębie tego szlaku dotyczą około 30–50% nowotworów złośliwych stercza[210][211]. Regulacja w górę szlaku PI3K/Akt/mTOR często jest następstwem utraty funkcji PTEN[211].
PTEN jest fosfatazą lipidową, która reguluje w dół szlak PI3K/Akt/mTOR. PTEN pełni rolę genu supresorowego, reguluje podziały komórkowe i w przypadku zatrzymania cyklu komórkowego przez RB posiada zdolność skierowania komórki na szlak apoptozy. Utrata PTEN wiąże się z nadmierną aktywacją szlaku PI3K/Akt/mTOR, co skutkuje zwiększoną proliferacją komórek, opornością na apoptozę i nasiloną angiogenezą[210][211]. Delecja genu PTEN jest rozpoznawana w 30% miejscowo ograniczonych przypadków raka gruczołu krokowego, a w 5–10% przypadków są obecne mutacje inaktywujące ten gen. W zaawansowanej chorobie odsetki te rosną[199]. Innym mechanizmem aktywacji szlaku PI3K/Akt/mTOR są amplifikacja genu i mutacje punktowe genu PIK3CA kodującego podjednostkę PI3K[199][212]. Mutacje aktywujące PIK3CA i mutacje PTEN są wzajemnie wykluczające, co potwierdza podobny efekt obu zmian cytogenetycznych[199].
- RB
Ważnym elementem karcynogenezy raka gruczołu krokowego jest inaktywacja genu supresorowego RB1 odpowiedzialnego za kontrolę proliferacji i progresji cyklu komórkowego[199]. W 1–14% przypadków raka gruczołu krokowego stwierdza się mutację RB1, a w 5–23% utratę tego genu[212]. Białko RB w przypadku uszkodzenia DNA poprzez związanie i zablokowanie czynników transkrypcyjnych należących do rodziny E2F, powoduje zablokowanie przejścia cyklu komórkowego z fazy G1 i przejściu w fazę S[213][214]. Zatem dysfunkcja RB związana ze zmianami cytogenetycznymi prowadzi do nasilenia proliferacji komórek nowotworowych[213]. Utrata funkcji RB ułatwia rozwój fenotypu oporności na kastrację, ponieważ gen receptora androgenowego jest genem docelowym E2F1[214][199].
- c-MYC
c-MYC jest czynnikiem transkrypcyjnym regulującym wzrost komórki, proliferację, progresję cyklu komórkowego, różnicowanie, a także wpływa na oporność na apoptozę[215]. W karcynogenezie raka gruczołu krokowego pełni rolę protoonkogenu[216]. Mutację c-MYC stwierdza się w 2–20% przypadków raka[212].
- p53
p53 jest genem supresorowym pełniącym kluczową rolę w utrzymywaniu stabilności genetycznej. Białko p53 posiada zdolność zatrzymania cyklu komórkowego, naprawy materiału genetycznego i skierowania komórki na szlak apoptozy[212][217]. W ograniczonych do gruczołu przypadkach raka prostaty mutacja p53 pojawia się w 10–20% guzów, jednak w chorobie z przerzutami dotyczy już 40% przypadków[218]. Utrata funkcji p53 zwiększa inwazyjność nowotworu i jego zdolność do wytwarzania przerzutów[217].
- CDKN1B (p27)
CDKN1B (p27) jest genem supresorowy kodującym białko p27 będące inhibitorem kinazy zależnej od cyklin[197]. Utrata funkcji tego genu jest stwierdzana w 23% miejscowo ograniczonych przypadków raka stercza i blisko 50% przypadków tego nowotworu w stadium rozsiewu[218].
- EZH2
EZH2 współtworzy kompleks PRC2, który poprzez metylację histonu H3 prowadzi do wyciszania genów zaangażowanych w różnicowanie[187]. W raku stercza EZH2 ulega nadmiernej ekspresji, co skutkuje wyciszaniem ekspresji wielu genów, jednak EZH2, obok osłabiania ekspresji niektórych genów, może wzmacniać ekspresję innych genów[219]. Wiadomo, że EZH2 jest konieczny do ekspresji genów związanych z E2F (szlak RB-E2F) nasilających proliferację[220][221]. Innym celem może być gen supresorowy Ink4a/Arf, szlak E-kadheryny oraz FOXC1[222][223][224][220].
- FOXA1
FOXA1 należy do podrodziny czynników transkrypcyjnych FOX, które wpływają na ekspresję genów regulujących cykl komórkowy, apoptozę, naprawę materiału genetycznego, metabolizm glukozy[225]. Obserwuje się wysoki poziom ekspresji FOXA w nieprzerzutowym raku gruczołu krokowego i silną ekspresję w przerzutowym opornym na kastrację raku stercza[226][227][228].
FOXA1 wpływa na wiązanie receptora androgenowego z jego miejscami wiązania w obrębie chromatyny[229][230]. W raku gruczołu krokowego FOXA1 może zarówno promować, jak i hamować wiązanie AR z miejscem wiązania, przez co znacząco wpływa na efekt działania AR i fenotyp komórki nowotworowej[231][229]. Prawdopodobnie FOXA1 przy pewnym poziomie ekspresji promuje różnicowanie komórki, a przy wyższym poziomie ekspresji sprzyja proliferacji komórek i przeżyciu[231].
- SPOP
SPOP koduje ligazę E3 ubikwityny zawierającą kulinę. Mutacja genu SPOP dotyczy około 5–15% nowotworów gruczołu krokowego[232][233][195]. Do mutacji najprawdopodobniej dochodzi we wczesnym etapie historii naturalnej nowotworu[195]. Mutacja SPOP występuje jedynie w przypadków nowotworów bez zmian w obrębie ETS[233][195].
W konsekwencji mutacji SPOP dochodzi do upośledzenia degradacji białek, w tym onkoprotein biorących udział w karcynogenezie takich jak AR i ERG[195][234][235]. Badania sugerują aktywację sygnalizacji AR i aktywację szlaku PI3K/Akt/mTOR[236]. SPOP bierze udział w procesie naprawy przerwanej nici DNA i mutacja tego genu powoduje niestabilność genetyczną[237][195].
- CHD1
CHD1 koduje białko o zdolności do remodelingu chromatyny i biorące udział w naprawie materiału genetycznego[212][195][238]. Mutacja pojawia się w 15–27% przypadków raka gruczołu krokowego[238], mutacje CHD1 są w znacznym stopniu powiązane z delecją SPOP[196]. W jej konsekwencji dochodzi do niestabilności genetycznej[238].
- SPINK1
Nadekspresja genu SPINK1 jest obecna w 10% nowotworów gruczołu krokowego, występuje ona wyłącznie w guzach bez zmian ETS i współwystępuje ona z mutacjami SPOP i CHD1[196][195].
Rozpoznanie

Większość przypadków raka gruczołu krokowego jest rozpoznawana w stadium bezobjawowym. Zmiany w gruczole krokowym stwierdzone w badaniu przez odbyt oraz nieprawidłowe stężenie PSA są podstawowymi przesłankami do wykonania przezodbytniczej biopsji stercza, podczas której pobiera się materiał tkankowy do badania histopatologicznego, które jest podstawą rozpoznania raka gruczołu krokowego[239].
Badanie per rectum
Część raków gruczołu krokowego może zostać wykryta podczas badania palcem przez odbytnicę[184]. Jego wartość diagnostyczna jest jednak ograniczona a skuteczność zależy od doświadczenia badającego[184]. Większość raków gruczołu krokowego jest umiejscowiona w strefie obwodowej i guzy mogą być rozpoznane przy objętości przekraczającej 0,2 ml[136]. Badanie przez odbyt (niezależnie od stężenia PSA) umożliwia wykrycie około 18% nowotworów stercza[240][136]. Przy wartości stężenia PSA poniżej 2 ng/ml badanie ma dodatnią wartość predykcyjną wynoszącą 5–30%[241][136]. Nieprawidłowy wynik badania przez odbyt przemawia za koniecznością wykonania biopsji[136].
Swoisty antygen sterczowy (PSA)
Swoisty antygen sterczowy (PSA) jest proteazą serynową wytwarzaną przez nabłonek gruczołu krokowego, który jest obecny w wydzielinie gruczołu krokowego[242]. Wzrost jego stężenia jest następstwem uszkodzenia warstwy podstawnej komórek nabłonka gruczołowego i błony podstawnej, a w konsekwencji jego przechodzenia do krwiobiegu[17]. PSA jest stosowany w diagnostyce i monitorowaniu leczenia raka gruczołu krokowego[243].
PSA jest markerem swoistym dla stercza, jednak nieswoistym dla samego raka gruczołu krokowego. Wzrost stężenia PSA, poza rakiem stercza, stwierdza się również w chorobach nienowotworowych, w tym w łagodnym rozroście gruczołu krokowego, zapaleniu gruczołu krokowego lub po zabiegach w zakresie stercza, w tym również po cewnikowaniu pęcherza moczowego[17]. Większość chorych (nawet 75%) z podwyższonym stężeniem PSA nie choruje na raka gruczołu krokowego, a jest ono związane z łagodnym rozrostem gruczołu krokowego lub jego zapaleniem[244].
Wartości referencyjne stężenia PSA zostały ustalone arbitralnie, za górną granicę normy przyjmuje się stężenie 4 ng/ml, a wartości 4–10 ng/ml pozostają „diagnostyczną szarą strefą”[242]. Zależność stężenia PSA od ryzyka obecności nowotworu złośliwego stanowi pewną ciągłość, w którym wyższe stężenie koreluje z wyższym prawdopodobieństwem obecności gruczolakoraka stercza[245][14]. Przy stężeniu 4–10 ng/ml prawdopodobieństwo stwierdzenia nowotworu podczas biopsji wynosi 20–30%, przy stężeniu powyżej 10 ng/ml takie ryzyko wynosi ponad 60%[14]. Wysokie stężenia często korelują z wyższym zaawansowaniem[242][243]. Z drugiej strony u części chorych rak stercza przebiega bez wzrostu stężenia PSA[245].
Prawdopodobieństwo występowania istotnego klinicznie RGK przy niskim PSA[246]
| PSA w ng/ml | Ryzyko występowania raka gruczołu krokowego (%) |
| 0,0 – 0,5 | 6,6 |
| 0,6 – 1,0 | 10,1 |
| 1,1 – 2,0 | 17,0 |
| 2,1 – 3,0 | 23,9 |
| 3,1 – 4,0 | 26,9 |
Wartość stężenia PSA rośnie wraz z wiekiem, co jest związane ze zwiększeniem objętości stercza z powodu łagodnego rozrostu gruczołu krokowego[14]. Zatem aby poprawić swoistość badania, zaproponowano wartości referencyjne uwzględniające wiek badanego[242]. Wówczas w tej metodzie górna granica normy stężenia PSA dla mężczyzn w wieku 40–49 lat wynosi poniżej 2,5 ng/ml, dla mężczyzn w wieku 50–59 lat poniżej 3,5 ng/ml, w wieku 60–69 lat poniżej 4,5 ng/ml i w wieku 70–79 lat poniżej 6,5 ng/ml[247][14].
Inną metodą mającą na celu poprawę swoistości PSA jest określenie współczynnika gęstości PSA (PSA density, PSAD), który jest stosunkiem stężenia PSA do objętości stercza zmierzonego podczas ultrasonografii przezodbytniczej lub za pomocą rezonansu magnetycznego[248][243]. Rak stercza wiąże się z wyższym współczynnikiem gęstości PSA w porównaniu do łagodnego rozrostu gruczołu krokowego[248]. Metoda pozwala poprawić swoistość badania PSA, jednak nie ustalono optymalnej wartości punktu odcięcia[249][242].
W surowicy 70–90% krążącego PSA jest związanych z inhibitorami proteaz takimi jak α1-antychymotrypsyna i α2-makroglobulina, pozostała część jest niezwiązana i określa się ją wolnym PSA[242]. W raku stercza stosunek wielkości frakcji wolnego PSA (fPSA) do całkowitego (tPSA) jest niższy niż w łagodnym rozroście gruczołu krokowego[243]. Ocena stosunku wolnego PSA (fPSA) do całkowitego (tPSA) u chorych ze stężeniem PSA 4–10 ng/ml i ujemnym badaniem przez odbyt pozwala oszacować ryzyko obecności raka gruczołu krokowego[250]. Przyjmując wartość odcięcia 25% (0,25) stosunku fPSA do tPSA, można uniknąć około 20% niepotrzebnych biopsji[251][242]. W jednym badaniu klinicznym 56% mężczyzn z wynikiem fPSA/tPSA poniżej 10% (0,1) chorowało na raka gruczołu krokowego, podczas gdy przy wartości tego stosunku powyżej 0,25 rak był wykrywany u 8% chorych[251][250][242]. Badanie nie ma zastosowania przy wartościach PSA powyżej 10 ng/ml oraz w przypadku już ustalonego rozpoznania raka gruczołu krokowego[250].
Kolejną metodą mającą na celu zwiększenie wartości predykcyjnej badania PSA jest ocena czasu podwojenia stężenia PSA (PSA doubling time, PSADT). Opiera się ona na szybszym wzroście stężenia PSA w określonym odstępie czasu u osób chorych na raka stercza niż u mężczyzn bez raka, również w przypadku nieprzekraczania wartości stężenia PSA uważanych za górną granicę normy[252]. Metoda wymaga kilku pomiarów w odstępach 1–2 letnich[242]. Wadą metody są związane z prawidłowo występującymi różnicami stężenia PSA, co przyczynia się do licznych wyników fałszywie dodatnich[252].
PCA3
W wydzielinie gruczołu krokowego mogą być obecne komórki nowotworowe, które mogą zostać wykryte za pomocą markerów molekularnych[252]. W raku gruczołu krokowego w moczu może być wykrywany niekodujący mRNA genu PCA3 (prostate cancer gene 3), który ulega znacznej nadekspresji[242]. PCA3 jest oznaczany z osadu moczu pobranego po masażu gruczołu krokowego wykonanego w trakcie badania przez odbyt[250][242]. Marker jest przydatny w kwalifikacji mężczyzn z podwyższonym stężeniem PSA po ujemnej biopsji stercza do kolejnej biopsji gruczołu krokowego[252][250]. Badanie PCA3 wykazuje wyższość nad oznaczeniem stosunku wolnego PSA do całkowitego w rozpoznawaniu raka stercza[250][253].
Biopsja

Rozpoznanie raka gruczołu krokowego wymaga wykonania przezodbytniczej biopsji stercza. Głównymi wskazaniami do wykonania takiej biopsji jest stwierdzenie nieprawidłowości w badaniu przez odbytnicę (badanie per rectum) oraz podwyższona wartość PSA[252][254].
Biopsja mapująca stercza
Biopsja mapująca stercza pod kontrolą ultrasonografii przezdodbytniczej (TRUS) jest standardowo stosowaną metodą diagnostyczną, która polega na wykonaniu określonej liczby nakłuć igłą biopsyjną w ustalone obszary stercza oraz dodatkowo we wszystkie wyczuwalne zmiany w badaniu przezodbytniczym lub uwidocznione radiologicznie[242]. Biopsja mapująca stercza faktycznie jest „biopsją ślepą”, co niesie pewne ograniczenia metody[255][256].
Typowo wykonuje się 10–12 biopsji za pomocą igieł wielkości 18G[242][244][257]. Pobiera się co najmniej 10 bioptatów w celu systematycznego odwzorowania gruczołu krokowego, a ponadto ze zmian wyczuwalnych palpacyjnie i uwidocznionych w TRUS lub MRI[252]. Opisuje się lokalizację i orientację każdego rdzenia, ponieważ umożliwia to określenie zasięgu nacieku nowotworowego w przypadku jego wykrycia[252][258]. Protokół z uzyskaniem 10–12 rdzenia w porównaniu do biopsji sekstantowej (pobranie 6 rdzeni tkankowych) zwiększa skuteczność wykrywania raka, zwiększa wartość ujemną predykcyjną badania i zmniejsza ryzyko konieczności powtarzania biopsji[258]. Jednak wzrost liczby biopsji powyżej 12 nie skutkuje już poprawą skuteczności[259][258][260].
Biopsja pod kontrolą rezonansu magnetycznego
Multiparametryczny rezonans magnetyczny jest metodą mającą wysoką czułość i swoistość w wykrywaniu ognisk nowotworowych w obrębie stercza[255]. Metoda ta pozwala na uwidocznienie podejrzanej zmiany i wykonanie biopsji celowanej[256].
Opracowano trzy techniki wykonania biopsji pod kontrolą MRI: biopsja pod bezpośrednią kontrolą MRI (MRI in-bore guided biopsy, MRI-IB-GB), biopsja pod kontrolą fuzji obrazów TRUS i mpMRI (biopsja fuzyjna, software fusion guided biopsy, FUS-GB) oraz biopsja kognitywna (fuzja obrazów MRI i TRUS w pamięci, cognitivefusion biopsy, COG-FB)[255][256]. Biopsja pod bezpośrednią kontrolą MRI jest technicznie skomplikowana i wymaga odpowiedniego sprzętu. Przed biopsją wykonuje się pierwsze badanie, w którym ustala się położenie zmiany. Następnie w oparciu o położenie zmiany, przy pomocy odpowiedniego oprogramowania, ustawia się prowadnicę dla igły biopsyjnej, a następnie wprowadza się igłę biopsyjną. Kolejne badanie rezonansu magnetycznego ma na celu potwierdzenia właściwego położenia igły w podejrzanej zmianie[261]. Ograniczeniem metody są ograniczona dostępność i wysokie koszty[256][262]. Fuzja obrazów MRI i TRUS w pamięci polega na ocenie przez badającego lekarza wyników wcześniej wykonanego badania MRI stercza i zapamiętaniu lokalizacji podejrzanych zmian, a następnie próbie ich odnalezienia w trakcie TRUS[256]. Fuzja obrazów MRI i TRUS za pomocą odpowiedniego oprogramowania (biopsja fuzyjna) polega na wcześniejszym wykonaniu rezonansu magnetycznego stercza oraz oznaczaniu podejrzanych obszarów stercza, a następnie połączeniu i naniesieniu w czasie rzeczywistym na obraz widoczny podczas wykonywania TRUS konturów stercza i obrysów podejrzanych ognisk. Po znalezieniu podejrzanego ogniska wykonuje się biopsję celowaną[256].
Obrazowanie multiparametrycznym rezonansem magnetycznym może być wykorzystane jako metoda poprawiająca skuteczność biopsji mapującej, wówczas w tej strategii diagnostycznej z pomocą rezonansu magnetycznego wykonuje się biopsje celowane zobrazowanych podejrzanych ognisk, a u mężczyzn z ujemnym wynikiem rezonansu magnetycznego biopsję mapującą[263]. Kilka badań sugeruje, że multiparametryczne obrazowanie rezonansem magnetycznym poprawia wykrywalność klinicznie istotnego raka stercza, jednak ten korzystny trend jest obserwowany przede wszystkim u mężczyzn poddanych kolejnej biopsji, a nie podczas pierwszorazowej biopsji[264][265][266][267][268][269]. Z kolei badania oceniające skuteczność wykrywania klinicznie istotnego raka stercza samodzielnej biopsji mapującej w porównaniu z kombinacją biopsji celowanej i biopsji mapującej u mężczyzn uprzednio nie poddawanych jeszcze biopsji dały sprzeczne wyniki[270][271][272][263]. W związku z tym EAU zleca wykonywanie mpMRI jedynie u chorych z klinicznym podejrzeniem raka gruczołu krokowego z ujemnym wynikiem biopsji przed wykonaniem kolejnej biopsji[273].
Drugą badaną strategią jest metoda, w której multiparametryczny rezonans magnetyczny stanowi jedyne kryterium kwalifikacji do biopsji i chorzy z uwidocznionymi podejrzanymi ogniskami są poddawani biopsji, a mężczyźni z prawidłowym wynikiem badania nie są poddawani biopsji[263]. Jednak bezpieczne odstępowanie od biopsji stercza u chorych z prawidłowym wynikiem mpMRI wymaga wypracowania metod określenia kategorii ryzyka występowania raka, co zwiększyłoby ujemną wartość predykcyjną badania (skuteczność badania wykluczającego chorobę). W celu identyfikacji takich grup chorych oceniono gęstość PSA oraz kalkulatory ryzyka, jednak konieczne są dalsze badania[263].
Ponowna biopsja stercza
Mężczyźni z ujemnym wynikiem biopsji stercza i klinicznym podejrzeniem raka stercza na podstawie nieprawidłowego wyniku badania przez odbyt, rosnących lub utrzymujących się podwyższonych wartości PSA, nieprawidłowego wyniku multiparametrycznego rezonansu magnetycznego stercza albo rozpoznaniem w poprzedniej biopsji atypowego rozrostu drobnozrazikowego (ASAP) albo rozległych ognisk śródnabłonkowej neoplazji stercza wysokiego stopnia (HGPIN) mogą wymagać wykonania ponownej biopsji[254]. Skuteczność diagnostyczna kolejnej biopsji zależy od typu i rodzaju wcześniej wykonywanej biopsji. Jednak ponad 90% przypadków raka stercza można rozpoznać już w samej biopsji sekstantowej[274][257], zatem przy obecnie stosowanych schematach jest bardzo mało prawdopodobne, by nie został on rozpoznany po wykonaniu dwóch biopsji[275][257].
U mężczyzn z podwyższonym ryzykiem raka stercza, po uprzedniej negatywnej biopsji można uzyskać dodatkowe informacje, wykonując testy w moczu (Progensa-PCA3 i SelectMDX DRE), testy w surowicy krwi (4Kscore i PHI) lub tkankowy test epigenetyczny (ConfirmMDx), który określa ilościowo poziom metylacji regionów promotorowych trzech genów w tkance prostaty. Rola tych testów w typowaniu do ponownej biopsji jest niepewna[276].
Biopsja saturacyjna
Biopsja saturacyjna polega pobraniu przynajmniej 20 rdzeni tkankowych z całego obszaru gruczołu krokowego[277][242][278]. Biopsja saturacyjna znajduje zastosowanie u chorych z klinicznym podejrzeniem raka stercza, u których podczas pierwszorazowej biopsji nie wykryto procesu nowotworowego[277]. Metoda poprawia wykrywalność raka stercza u chorych z ujemnymi wynikami wcześniejszych biopsji, jednak jako metoda pierwszorazowej biopsji nie poprawia wyników rozpoznawania raka[279][242].
Badania obrazowe
Diagnostyka obrazowa jest pomocna w rozpoznawaniu choroby oraz jest wykorzystywana w ocenie zaawansowania choroby i rozpoznawaniu jej nawrotu[280][281].
Ultrasonografia przezodbytnicza

Ultrasonografia przezodbytnicza (TRUS) jest najczęściej stosowaną metodą obrazującą gruczoł krokowy[282]. Metoda pozwala wizualizować anatomię gruczołu krokowego oraz określić poszczególne obszary stercza, co pozwala wykonać biopsję mapującą gruczołu krokowego[252][242].
Rak gruczołu krokowego w ultrasonografii przezodbytniczej najczęściej jest widoczny jako zmiana hipoechogeniczna (60% zmian), jednak nowotwór może być normoechogeniczny (blisko 40% zmian) lub hiperechogeniczny (1% zmian)[256]. Do zalet ultrasonografii przezodbytniczej należy możliwość obrazowania w czasie rzeczywistym i wysoka dostępność badania[282]. Jednak ultrasonografia przezodbytnicza wykazuje stosunkowo niską czułość w wykrywaniu raka gruczołu krokowego[282][256]. Ocenia się, że badanie wykazuje 50–60% dokładność diagnostyczną i jedynie 6% dodatnią wartość predykcyjną[283]. Jest to związane ze zmiennością obrazu ultrasonograficznego choroby, częstym izoechogenicznym obrazem oraz niską swoistością obserwowanych zmian echograficznych, ponadto obraz ultrasonograficzny często współwystępującego łagodnego rozrostu stercza może maskować zmiany złośliwe położone w centralnej części stercza[283]. Podobnie czułość (50–92%), swoistość (46–91%) i dokładność diagnostyczna (58–86%) oceny zaawansowania miejscowego jest słaba[283]. Metoda ma ograniczoną skuteczność oceny szerzenia się raka poza stercz i nie odgrywa istotnej roli w rozpoznaniu obecności rozsiewu[282]. Nowe techniki ultrasonografii takie jak ultrasonografia z zastosowaniem środków kontrastowych czy sonoelastografia pozostają w trakcie badań klinicznych i nie są rutynowo stosowane[263].
Multiparametryczny rezonans magnetyczny
Rezonans magnetyczny (MRI) umożliwia dobrą wizualizację gruczołu krokowego oraz otaczających tkanek[284]. Rezonans magnetyczny stanowi podstawową metodę diagnostyki obrazowej raka gruczołu krokowego[285]. Ze względu na różne właściwości poszczególnych technik anatomicznych i funkcjonalnych rezonansu magnetycznego w celu poprawy dokładności rozpoznania raka stercza łączy się je w technikę wieloparametrową – multiparametryczny rezonans magnetyczny (mpMRI)[285]. W multiparametrycznym rezonansie magnetycznym łączy się sekwencje anatomiczne, przede wszystkim skany T2-zależne, z przynajmniej dwoma sekwencjami funkcjonalnymi: obrazowaniem rezonansu magnetycznego zależnym od dyfuzji (DWI), obrazowaniem dynamicznym ze wzmocnieniem kontrastowym (DCE) lub spektroskopią rezonansu magnetycznego (MRS)[286].
Multiparametryczny rezonans magnetyczny wykazuje wysoką czułość w wykrywaniu nowotworów o złośliwości w skali Gleasona większej niż lub równej 7[263]. Ocenia się, że czułość w wykrywaniu guzów wielkości poniżej 0,5 ml o złośliwości 7 i powyżej 7 wynoszą odpowiednio 63% i 80%, zmian 0,5–2 ml odpowiednio 82–88% i 93% oraz zmian powyżej 2 ml 97% i 100%[287][263]. Badanie cechuje się wysoką wartością predykcyjną dodatnią i wysoką wartością predykcyjną ujemną[263]. W związku z tym mpMRI jest często wykonywany jeszcze przed biopsją gruczołu krokowego[263].
W obrazach T2-zależnych rak gruczołu krokowego w strefie obwodowej prezentuje się jako okrągłe, słabo odgraniczone ognisko o niskim sygnale, które kontrastuje z hiperintensywnym obszarem prawidłowej tkanki stercza[286]. Jednak podobny obraz mogą naśladować zapalenie stercza, guzki hiperplastyczne, blizny, obszary zawału i krwotok[285]. Nowotwory strefy przejściowej zwykle są nieodróżnialne od otaczającego gruczołu[285]. W strefie przejściowej w obrazach T2-zależnych rak może dawać obraz homogennej zmiany o niewyraźnych granicach o niskiej intensywności sygnału, jednak nowotwory tej strefy stercza często są trudne do odróżnienia od występującego w tej strefie łagodnego rozrostu gruczołu krokowego[286][285].
Szerzenie się pozatorebkowe jest widoczne jako obecność zmiany o pośrednim sygnale penetrującej przez torebkę gruczołu krokowego, w przypadku mniej nasilonego nacieku inwazja torebki stercza może objawiać się jako przyleganie na dużym obszarze guza nowotworowego do torebki, jako pogrubienie, guzkowatość lub wybrzuszenie torebki lub jej nieregularność. Metoda pozwala na ocenę naciekania pęcherzyków nasiennych, co jest widoczne jako obszar o niskiej intensywności sygnału, oraz naciek pęcherza moczowego prezentujący się jako obszar o pośredniej intensywności sygnału w obrazach T2-zależnych albo obszar w obrębie ściany narządu ulegający wzmocnieniu kontrastowemu[282].
Obrazowanie dyfuzyjne (DWI) jest techniką umożliwiacą ocenę przypadkowych ruchów Browna cząstek wody w przestrzeni pozakomórkowej[286][288]. Rak stercza cechuje się wysoką gęstością komórkową lub obecnością gęsto upakowanych komórek nowotworowych, co przekłada się na zmianę stosunku objętościowego przestrzeni wewnątrzkomórkowych i zewnątrzkomórkowej, co prowadzi do ograniczenia dyfuzji i obniżenia wyliczonego pozornego współczynnika dyfuzji (ADC)[286]. Rak stercza najczęściej w DWI w porównaniu do prawidłowej tkanki wykazuje utrudnioną dyfuzję i na mapie ADC jest widoczny jako obszar hipointensywny[288]. Wadą metody jest stosunkowo słaba rozdzielczość przestrzenna[285]. Zmiany widoczne w DWI muszą być skorelowane z obrazami T1-zależnymi, T2-zależnymi i obrazowaniem z dynamicznym wzmocnieniem kontrastowym[288].
Obrazowanie z dynamicznym wzmocnieniem kontrastowym (DCE) jest uzyskiwane po podaniu kontrastu gadolinowego i wykonaniu szybkiego skanu w obrazie T1-zależnym[288]. W tkance nowotworowej występuje zwiększona neoangiogeneza, patologiczne naczynia krwionośne są bardziej przepuszczalne, co pozwala środkowi kontrastowemu szybko ulegać dyfuzji do pozanaczyniowej przestrzeni zewnątrzkomórkowej[286]. Obrazy w DCE są analizowane ilościowo, półilościowo i jakościowo. Ocena ilościowa polega na wizualnej ocenie podejrzanych obszarów. Analiza półilościowa polega na ocenie szybkości wzmocnienia kontrastowego i wypłukania kontrastu poprzez wyznaczenie krzywej kinetyki zmian. W ocenie jakościowej poprzez zastosowanie farmakokinetycznego modelowania przedziałowego uwzględniającego stężenie środka kontrastowego i funkcję napływu tętniczego wyznacza się stałe czasu do szybkości napływu (K-trans) i wypłukiwania (kep) środka kontrastowego[289][286][288]. Umożliwia to wyznaczenie mapy K-trans i kep ułatwiającej rozpoznanie nieprawidłowości[288].
Wzmocnienie raka stercza jest w pewnym stopniu zmienne[282]. Rak gruczołu krokowego w DCE po podaniu kontrastu wykazuje wczesne wzmocnienie kontrastowe w porównaniu do otaczającej prawidłowej tkanki, co powinno odpowiadać zarejestrowanym zmianom w obrazach T2-zależnych lub DWI[288][289].
Spektroskopia rezonansu magnetycznego (MRS) opiera się o ocenę stężeń określonych kluczowych metabolitów w tkance gruczołu krokowego, w tym choliny, kreatyniny i cytrynianów[290]. W prawidłowej tkance gruczołu krokowego obserwuje się względnie wysokie stężenie cytrynianu, który w tkance nowotworowej jest obecny w niższym stężeniu. Z kolei w komórkach nowotworowych raka stercza obserwuje się wyższe poziomy choliny. Typowo w raku stercza stwierdza się podwyższony stosunek stężenia choliny do cytrynianu[290][286]. Ze względów technicznych (trudności z odróżnieniem piku dla choliny i kreatyniny) zwykle jest oceniany stosunek choliny z kreatyniną do cytrynianu[286].
Multiparametryczny rezonans magnetyczny jest najbardziej przydatną metodą diagnostyki w ocenie zaawansowania klinicznego. Metoda wykazuje wysoką swoistość, ale stosunkowo niską czułość w wykrywaniu naciekania poza torebką gruczołu krokowego i naciekania pęcherzyków nasiennych (zaawansowanie T3)[291]. Ocenia się, że mpMRI wykazuje 57% czułość i 91% swoistość w wykrywaniu naciekania poza torebkę stercza oraz 58% czułość i 96% swoistość w rozpoznawaniu naciekania pęcherzyków nasiennych[292][291]. Zastosowanie aparatów 3T oraz DWI poprawia skuteczność wykrywania naciekania pozastercza[291].
Tomografia komputerowa

.jpg)
Tomografia komputerowa (TK) znajduje zastosowanie w ocenie zaawansowania raka gruczołu krokowego. Pozwala na rozpoznawanie przerzutów do narządów klatki piersiowej, jamy brzusznej, miednicy, kości czy węzłów chłonnych[282]. Jednak tomografia komputerowa nie znajduje większego zastosowania w ocenie zmian wewnątrz samego gruczołu krokowego[282].
Scyntygrafia

Scyntygrafia z zastosowaniem 99mTc jest szeroko wykorzystywana w diagnostyce przerzutów raka gruczołu krokowego do kości[293]. Radiofarmaceutyk zawierający 99mTc preferencyjnie gromadzący się w kościach emituje promieniowanie gamma, które jest wykrywane przez kamerę gamma[282]. Metoda wykazuje 79% czułość i 82% swoistość w wykrywaniu rozsiewu do kości oraz 59% czułość i 75% swoistość w rozpoznawaniu poszczególnych przerzutów[294][293].
Tomografia emisyjna pojedynczych fotonów (SPECT) jest wykonywana na podobnej zasadzie, przy czym uzyskuje się projekcje z wielu kątów, co pozwala uzyskać obrazy przestrzenne[282]. W porównaniu do scytygrafii SPECT wykazuje większą czułość i swoistość w wykrywaniu przerzutów do kości[295].
Pozytonowa tomografia emisyjna

Pozytonową tomografię emisyjną (PET) wykorzystuje się w ocenie zaawansowania raka gruczołu krokowego[296].
PET ze znakowaną choliną (11C-cholina i 18F-cholina) wykazuje większą czułość i swoistość w ocenie obecności przerzutów do kości i do węzłów chłonnych niż scyntygrafia czy mpMRI[296]. W badaniu porównującym skuteczność PET ze znakowaną choliną ze scyntygrafią i tomografią komputerową brzucha wykazano wyższą czułość i swoistość PET ze znakowaną choliną w rozpoznawaniu przerzutów do kości niż scyntygrafii wynoszącą odpowiednio 100% i 86,4% dla PET oraz 90% i 77,2% dla scyntygrafii[297][296]. W innej metaanalizie czułość PET ze znakowaną choliną w wykrywaniu przerzutów do węzłów chłonnych została oceniona na 62%, a swoistość na 92%[298]. Ze względu na ograniczoną dostępność PET ze znakowaną choliną oraz niejasną efektywność kosztową rutynowymi metodami oceny obecności przerzutów w kościach pozostają scyntygrafia oraz tomografia komputerowa[296]. Metoda wykazuje umiarkowaną skuteczność w wykrywaniu przerzutów do węzłów chłonnych, dlatego nie jest zalecana w rutynowej ocenie rozsiewu do węzłów chłonnych[296].
Cholina, jako marker proliferacji błon komórkowych, jest znakowana radioaktywnym fluorem-18 (18F) lub węglem-11 (11C). W metaanalizach wykazano, że czułość i swoistość radiocholin w wykrywaniu ognisk wznowy PCa wynosi odpowiednio 86–89% i 89–93%[299]. Co więcej, PET/CT z radiocholiną może uwidocznić mnogie ogniska przerzutowe w kościach u pacjentów z pojedynczym przerzutem widocznym w scyntygrafii. Czułość PET/CT z radiocholiną w istotny sposób zależy jednak od stężenia PSA. U pacjentów ze wznową biochemiczną choroby (BCR) po radykalnej prostatektomii badanie daje dodatni wynik zaledwie u 5–24% pacjentów z PSA <1,0 ng/ml, u pacjentów z PSA >5,0 ng/ml odsetek ten wzrasta do 67–100%[300]. Zaleca się, aby obrazowanie PET/CT z radiocholiną przeprowadzać tylko u tych chorych, u których stężenie PSA przekracza 1,0 ng/ml[301]
PET z zastosowaniem znakowanego fluorku sodu (18F-NaF PET) znajduje zastosowanie w diagnostyce zmian przerzutowych do kości. Radioaktywny fluor poprzez przekształcenie hydroksyapatytu we fluoroapatyt jest wbudowywany do kości[296]. Metoda wykazuje wyższą czułość i swoistość w porównaniu do scyntygrafii kości i SPECT[302][296]. 18F-NaF PET nie nadaje się do oceny zmian w obrębie węzłów chłonnych, ponadto zastosowanie ograniczają niewielka dostępność i wysoki koszt[293].
Ze względu na niski wychwyt glukozy przez raka gruczołu krokowego zastosowanie FDG-PET jest mocno ograniczone[296].
W badaniu PET, oprócz wymienionych powyżej ligandów (FDG, cholina, octan) kolejnym znacznikiem w obrazowaniu czynnościowym jest ligand antygenu błony komórek stercza (prostate-specific membrame antygen – PSMA). PSMA jest glikoproteiną przezbłonową o aktywności enzymatycznej, która ulega ekspresji na powierzchni komórek prostaty, a także ślinianek, nerek, komórek nerwowych, dwunastnicy i jelita grubego. Ekspresja PSMA w komórkach PCa zwiększa się 100 do 1000-krotnie w porównaniu do zdrowych tkanek, co (pomimo braku specyficzności) umożliwia jego wykorzystanie w obrazowaniu PET/CT raka stercza. Dzięki temu badanie to w sposób wybiórczy identyfikuje komórki nowotworowe raka prostaty już w bardzo wczesnych etapach wznowy lub przerzutów[303].
Pozytonowa tomografia emisyjna (PET/CT) celowana na antygen błonowy gruczołu krokowego (PSMA) ma już ugruntowaną pozycję w obrazowaniu raka gruczołu krokowego, zwłaszcza u pacjentów ze wznową biochemiczną (biochemical relapse – BCR) choroby. W tej grupie chorych radioznaczniki celowane na PSMA cechują się największą czułością i swoistością. Badania dowodzą, że PET/CT z wykorzystaniem 68Ga-PSMA-11 (najlepiej obecnie przebadany znacznik nakierowany na antygen błonowy gruczołu krokowego) wykrywa miejsce wznowy PCa nawet u 50–58% pacjentów z BCR i bardzo małymi stężeniami PSA (<0,5 ng/ml)[304]. Wykazano, że wynik badania PET/CT ze znacznikiem 68Ga-PSMA-11 może zmienić sposób leczenia nawet u 76% pacjentów. Obrazowanie z użyciem radiofarmaceutyków wyznakowanych galem 68 (68Ga) ma jednak dość istotne wady. 68Ga-PSMA-11 wydalany jest przez układ moczowy, w związku z tym radioaktywny mocz gromadzący się w pęcherzu moczowym utrudnia ocenę gruczołu krokowego, loży po jego usunięciu i przylegających struktur. Dodatkowo 68Ga jest izotopem o dość ograniczonej dostępności. Z generatora 68Ge/68Ga można uzyskać zaledwie 2–4 dawki izotopu na dobę, a jego krótki okres półtrwania (ok. 68 min) właściwie uniemożliwia jego transport na dalsze odległości, w związku z czym obrazowanie z 68Ga mogą wykonywać tylko jednostki posiadające generatory[305].
Okres półtrwania fluoru 18 (18F) wynosi natomiast około 110 min, dlatego może być on produkowany w cyklotronach w dużych ilościach, a następnie transportowany nawet do oddalonych jednostek badawczych. Obrazy PET z radioznacznikami oznakowanymi 18F cechują się również dużo lepszą rozdzielczością w porównaniu z 68Ga[306]. Korzystny profil farmakokinetyczny i farmakodynamiczny radioznaczników celowanych na PSMA oznakowanych fluorem 18 wyjaśnia ich coraz powszechniejsze stosowanie w obrazowaniu PET/CT.
Radioznacznik 18F-PSMA-1007 został opracowany i zsyntetyzowany po raz pierwszy w 2016 r. przez badaczy w Heidelbergu w Niemczech. Pod względem chemicznym jest to kwas (3S,10S,14S)-1-(4-(((S)-4-karboksy-2-((S)-4-karboksy-2-(6-fluoronikotynamido)butanamido)butanamido)metylo) fenylo)-3-naftalen-2-ylmetylo)-1,4,12-triokso-2,5,11,13-tetraazaheksaadekano-0,14,16-trikarboksylowy[307].
18F-PSMA-1007 (podobnie jak większość badanych obecnie znaczników celowanych na PSMA) należy do tzw. drobnocząsteczkowych inhibitorów PSMA, które łączą się z centrum aktywnym enzymu za pomocą grupy mocznikowej. Po przyłączeniu ligandu (znacznika) do enzymu (PSMA) dochodzi do internalizacji kompleksu ligand–enzym i jego uwięzieniu wewnątrz komórki. Dzięki temu komórki wykazujące nadekspresję antygenu PSMA (komórki raka gruczołu krokowego) są widoczne w badaniu PET/CT jako miejsca zwiększonego wychwytu znacznika (aktywne metabolicznie)[308].
18F-PSMA-1007 spośród pozostałych drobnocząsteczkowych inhibitorów PSMA zdecydowanie wyróżnia korzystny sposób wydalania. 18F-PSMA-1007 wydala się z moczem, ale w wyniku czasowej retencji znacznika w miąższu nerek jest filtrowany do moczu z opóźnieniem. Dzięki temu w pęcherzu moczowym rejestrowane są śladowe ilości promieniowania – po 0–2 h od podania dawki jest to około 1,2% dawki podanej, a po 4–6 h około 0,5%. Małe gromadzenie znacznika w pęcherzu moczowym umożliwia dokładną ocenę gruczołu krokowego i otaczających tkanek, co w przypadku innych znaczników z tej grupy obarczone jest dużym ryzykiem błędu[309].
Wykazano, że obrazowanie PET/CT z tym znacznikiem uwidacznia zmiany patologiczne u 81–95% badanych. Widoczne są nawet zmiany o bardzo małych rozmiarach, np. przerzuty w niepowiększonych węzłach chłonnych. Tak jak w przypadku radiocholin i pozostałych znaczników celowanych na PSMA, czułość PET/CT z 18F-PSMA-1007 istotnie zależy od stężenia PSA pacjenta[310]. Przy stężeniu >2 ng/ml wynosi 94–100%. U pacjentów z bardzo małymi stężeniami PSA (<0,5 ng/ml) wykrywalność jest nadal stosunkowo duża i sięga 61–85%[311]. Dla porównania, radiocholiny (najczęściej obecnie stosowane znaczniki w diagnostyce BCR) przy tak małym stężeniu PSA cechują się wykrywalnością rzędu 12%[312].
Grupa drobnocząsteczkowych inhibitorów PSMA cały czas powiększa się o nowe związki, które potencjalnie w przyszłości mogą posłużyć do obrazowania PCa. Jak do tej pory 18F-PSMA-1007 i 18F-DCFPyL dominują w tej grupie zarówno jeśli chodzi o liczbę przebadanych pacjentów, jak i wartość diagnostyczną badania. Oba związki wykazują właściwie identyczną czułość. 18F-PSMA-1007 pozwala jednak na lepszą ocenę miednicy mniejszej ze względu na małe gromadzenie znacznika w moczu, 18F-DCFPyL natomiast w mniejszym stopniu gromadzi się w wątrobie, dlatego wydaje się znacznikiem odpowiedniejszym dla pacjentów z zaawansowaną chorobą i możliwością przerzutów do tego narządu[313].
Należy zwrócić uwagę, że rozmiary wykrywanych zmian są coraz mniejsze, co znacznie utrudnia ich biopsję i późniejszą analizę mikroskopową. W badaniu opublikowanym przez Giesela i wsp., w którym zmiany uwidocznione w 18F-PSMA-1007 PET/CT były weryfikowane histopatologicznie, wykazano, że korelacja pomiędzy dodatnim wynikiem PET/CT a obecnością komórek PCa w preparatach jest bardzo duża – czułość obrazowania wyniosła 97,4%[307].
Ocena zaawansowania choroby
Zaawansowanie choroby jest oceniane na podstawie wyniku badania przez odbyt, stężenia PSA oraz wyników badań obrazowych[314]. U większości chorych w momencie rozpoznania raka gruczołu krokowego nie występują przerzuty, dlatego u nich wykonywanie badań obrazowych mających na celu ocenę zaawansowania choroby nie jest konieczne[284]. U chorych z niskim ryzykiem (cT1a–T2a i GS <7 i PSA <10 ng/ml) nie ma konieczności wykonywania badań obrazowych celem oceny obecności przerzutów. U chorych o pośrednim (cT2b lub GS 7 lub PSA 10–20 ng/ml) lub wysokim ryzyku (≥T2c lub GS >7 lub PSA >20 ng/ml) w celu oceny miejscowego zaawansowania wykonuje się multiparametryczny rezonans magnetyczny gruczołu krokowego, a w celu oceny obecności przerzutów wykonuje TK albo MRI jamy brzusznej oraz scyntygrafię kości albo SPECT[315].
Badanie histopatologiczne
Badanie histopatologiczne materiału tkankowego uzyskanego drogą biopsji umożliwia ostateczne rozpoznanie raka gruczołu krokowego[273]. Materiał tkankowy po odpowiednim przygotowaniu i wybarwieniu jest oceniany pod mikroskopem. W ramach wyniku badania histopatologicznego opisuje się rozpoznanie typu histopatologicznego, stopień złośliwości według klasyfikacji Gleasona i powierzchnię utkania nowotworowego w stosunku do powierzchni bez nowotworu oraz zawiera informację o ewentualnym szerzeniu się poza gruczoł krokowy, naciek pęcherzyków nasiennych, naciek struktur naczyniowych i nerwów[273].
Zaawansowanie kliniczne
Zaawansowanie raka gruczołu krokowego jest oceniane w oparciu o klasyfikację TNM[316][317].
| Guz pierwotny – cecha T | |
| Tx | Nie można ocenić guza pierwotnego |
| T0 | Nie stwierdza się guza pierwotnego |
| T1 | Guz niewykrywalny w badaniu palpacyjnym lub obrazowym |
| T1a | Guz wykryty przypadkowo w badaniu histopatologicznym stanowiący ≤5% tkanki gruczołu krokowego |
| T1b | Guz wykryty przypadkowo w badaniu histopatologicznym stanowiący >5% tkanki gruczołu krokowego |
| T1c | Guz wykryty podczas biopsji igłowej |
| T2 | Guz ograniczony do gruczołu krokowego |
| T2a | Guz zajmuje nie więcej niż połowę jednego płata |
| T2b | Guz zajmuje ponad połowę jednego płata, ale nie zajmuje obu płatów |
| T2c | Guz zajmuje oba płaty |
| T3 | Guz naciekający poza torebkę gruczołu krokowego |
| T3a | Naciekanie pozatorebkowe, w tym również mikroskopowe naciekanie szyi pęcherza |
| T3b | Naciekanie pęcherzyków nasiennych |
| T4 | Guz nieruchomy lub nacieka sąsiednie struktury inne niż pęcherzyki nasienne |
| Zajęcie okolicznych węzłów chłonnych – cecha N | |
| Nx | Nie można ocenić okolicznych węzłów chłonnych |
| N0 | Nie stwierdza się przerzutów w okolicznych węzłach chłonnych |
| N1 | Przerzuty w regionalnych węzłach chłonnych |
| N2 | Przerzuty w licznych węzłach chłonnych w miednicy mniejszej (podbrzusznych, zasłonowych, biodrowych zewnętrznych lub przedkrzyżowych) |
| N3 | Przerzut w węźle chłonnym biodrowym wspólnym |
| Przerzuty odległe – cecha M | |
| Mx | Nie można określić obecności przerzutów odległych |
| M0 | Nie stwierdza się przerzutów odległych |
| M1 | Obecne przerzuty odległe |
| M1a | Przerzuty do pozaregionalnych węzłów chłonnych |
| M1b | Przerzuty do kości |
| M1c | Przerzuty o innej lokalizacji |
Leczenie
Postępowanie terapeutyczne po rozpoznaniu raka gruczołu krokowego zależy od stopnia zaawansowania choroby, ocenionej kategorii ryzyka choroby, obciążeń chorobami współistniejącymi, wieku chorego, przewidywanego czasu przeżycia, preferencji chorego i oceny wpływu leczenia na jakość życia chorego[319][320][321]. Według zaleceń EAU[322] i opracowania AOTMiT[323] w wyborze postępowania terapeutycznego należy rozpatrywać niżej przedstawione opcje:
1. w RGK niskiego ryzyka:
1.1. aktywny nadzór (dla pacjentów z oczekiwną długością przeżycia >10 lat),
1.2. baczną obserwcję (dla pacjentów z oczekiwaną długością przeżycia >10 lat),
1.3. leczenie chirurgiczne:
1.3.1. prostatektomię radykalną,
1.3.2. orchiektomię.
1.4. radioterapię:
1.4.1. z modulacją intensywności,
1.4.2. hipofrakcjonowaną z modulacją intensywności,
1.4.3. brachyterapię LDR,
1.4.4. brachyterapię poprzedzoną ADT.
2. w RGK średniego ryzyka:
2.1. baczną obserwcję (leczenie paliatywne),
2.2. leczenie chirugiczne:
2.2.1. prostatektomię radykalną z limfadenektomią,
2.2.2. orchiektomię,
2.3. radioterapię:
2.3.1. EBRT + ADT neoadjuwantową krótkoterminową,
2.3.2. EBRT + ADT adjuwantową krótkoterminową,
2.3.3. brachyterapię LDR,
2.3.4. brachyterapię LDR + teleradioterapię,
2.3.5. EBRT ze zwiększoną dawką.
2.4. farmakoterapię hormonalną:
2.4.1. neoadiuwantową, adjuwantową skojarzoną z radioterapią,
2.4.2. monoterapię (jeśli brak możliwości innego leczenia pacjenta objawowego).
3. w RGK wysokiego ryzyka:
3.1. baczną obserwację (leczenie paliatywne)
3.2. leczenie chirurgiczne:
3.2.1. prostatektomię radykalną z limfadenektomią,
3.2.2. orchiektomię
3.3. radioterapię:
3.3.1. EBRT + ADT neoadiuwantową długoterminową,
3.3.2. EBRT + ADT adiuwantową długoterminową,
3.3.3. EBRT + ADT + wzmocnioną brachyterapię (HDR/LDR),
3.3.4. brachyterapię + radioterapię,
3.3.5. brachyterapię LDR + teleradioterapię,
3.3.6. brachyterapię + ADT,
3.3.7. SBRT + ADT.
3.4. farmakoterapię hormonalną:
3.4.1. neoadjuwantową, adjuwantową skojazroną z radioterapią,
3.4.2. adjuwantową skojarzoną z chemioterapią,
3.4.3. monoterapię (jeśli brak możliwości innego leczenia pacjenta objawowego).
3.4.4. chemioterapię.
4. w lokalnie zaawansowanym RGK (z przerzutami do węzłów chłonnych):
4.1. baczną obserwcję (leczenie paliatywne),
4.2. leczenie chirurgiczne:
4.2.1. prostatektomię radykalną z limfadenektomią,
4.2.2. orchiektomię.
4.3. radioterapię:
4.3.1. EBRT + ADT neoadiuwantową długoterminową,
4.3.2. EBRT + ADT adiuwantową długoterminową,
4.3.3. IMRT/VMAT + ADT adjuwantową długoterminową,
4.3.4. hipofrakcjonowanie IMRT
4.4. farmakoterapię hormonalną:
4.4.1. neoadjuwantową, adjuwantową skojarzoną z radioterapią,
4.4.2. ADT adjuwantową,
4.4.3. monoterapię (jeśli brak możliwości innego leczenia pacjenta objawowego).
4.5. chemioterapię
5. w RGK z przerzutami odległymi:
5.1. baczną obserwcję (leczenie paliatywne),
5.2. radioterapię (jeśli mała liczba przerzutów),
5.3. farmakoterapię hormonalną:
5.3.1. kastrację + radioterapię,
5.3.2. kastrację hormonalną + radioterapię
5.3.3. kastrację + octan abirateronu + prednizon,
5.3.4. agonistów LHR + antyandrogeny,
5.3.5. kastrację + antyandrogeny,
5.3.6. monoterapię antyandrogenami.
5.4. chemioterapię:
5.4.1. monoterapię,
5.4.2. chemioterapię + hormonoterapię (przy chorobie nie leczonej hormonalnie),
6. w RGK opornym na kastrację:
– bez przerzutów:
6.1. monoterapię (enzalutamid lub apalutamid)
– z przerzutami:
6.2. ciągłe leczenie ADT jako leczenie pierwszego rzutu,
6.3. enzalutamid/abirateron + chemioterapię (docetaksel),
6.4. chemioterapię + enzalutamid/abirateron,
6.5. terapie przedłużające życie:
6.5.1. manipulację lekami (w zależności od powyżej stosowanymi),
6.5.2. radiofarmaceutyki w przerzutach do kości (rad 223, stront 89, samar 153).
Nie wszyscy chory z rozpoznaniem miejscowo ograniczonego raka gruczołu krokowego o niskim ryzyku od razu wymagają szybkiego wdrożenia leczenia przyczynowego choroby, ponieważ u większości chorych w tej grupie ryzyka nie rozwinie się choroba mająca wpływ na długość życia lub będąca powodem uciążliwych objawów – dlatego nie wszyscy chorzy z rakiem o niskim ryzyku odnoszą korzyść z leczenia[324]. U większości chorych w grupie niskiego ryzyka proponuje się aktywny nadzór polegający na monitorowaniu przebiegu choroby i ewentualnym wdrożeniu leczenia w przypadku rozpoznania jej progresji[325][326]. Innymi możliwościami w tej grupie chorych są teleradioterapia, brachyterapia albo radykalna prostatektomia[327]. U chorych z rozpoznaniem miejscowo ograniczonego raka gruczołu krokowego o pośrednim ryzyku wykonuje się prostatektomię albo radioterapię z pól zewnętrznych (EBRT) w skojarzeniu lub bez skojarzenia z ablacją androgenową (hormonoterapia) stosowaną przez okres 4–6 miesięcy, z brachyterapią lub bez niej, a u wybranych chorych z rakiem o niewielkiej objętości przydatna może być brachyterapia[328]. W leczeniu miejscowo ograniczonego raka o wysokim ryzyku stosuje się radioterapię z pól zewnętrznych stosowaną w skojarzeniu lub bez skojarzenia z ablacją androgenową stosowaną przez 2–3 lata, radioterapię z pól zewnętrznych stosowaną w skojarzeniu lub bez skojarzenia z brachyterapią, z lub bez połączenia z ablacją androgenową stosowaną przez 2–3 lata albo wykonuje się prostatektomię[328].
Leczenie choroby zaawansowanej z przerzutami opiera się na hormonoterapii[329][330]. Polega ona na kastracji chirurgicznej lub farmakologicznym zablokowaniu produkcji androgenów, nazywanym ablacją androgenową (ADT). W celu uzyskania ablacji androgenowej stosuje się agonisty lub antagonisty gonadoliberyny (LHRH, GnRH). Inną metodą jest użycie antyandrogenów (antagonistów receptora androgenowego)[331].
Biochemiczna lub radiologiczna progresja choroby, mimo uzyskania odpowiedniego stężenia testosteronu, jest związana z rozwojem raka opornego na kastrację (CRPC). W jego leczeniu stosuje się docetaksel, abirateron, enzalutamid, sipuleucel-T lub radioaktywny rad (223Ra), a wybór odpowiedniej metody jest oparty o cechy kliniczne choroby oraz stan chorego[332]. Podczas leczenia raka opornego na kastrację nadal konieczne jest utrzymywanie kastracyjnego stężenia testosteronu[333].
Aktywny nadzór
Rak gruczołu krokowego charakteryzuje się zróżnicowanym przebiegiem klinicznym, u części chorych choroba manifestuje agresywny przebieg, dający przerzuty i prowadzący do śmierci chorego, ale u znacznej części przebiega stosunkowo łagodnie. Zatem nie wszyscy chorzy z miejscowo ograniczonym rakiem gruczołu krokowego wymagają i odnoszą korzyści z leczenia, a jest ono konieczne u chorych z chorobą z cechami wskazującymi na jej agresywną historię naturalną[324]. Z kolei szerokie stosowanie oznaczenia stężenia PSA u mężczyzn bez jakichkolwiek objawów choroby spowodowało częste rozpoznawanie nieistotnych klinicznie przypadków raka gruczołu krokowego, czyli niemających wpływu na długość życia[334]. Aktywny nadzór polega na okresowym monitorowaniu przebiegu choroby u chorych z potencjalnie uleczalnym rakiem gruczołu krokowego w celu odroczenia leczenia radykalnego, aż do ewentualnego wykrycia bardziej agresywnego nowotworu lub progresji choroby[325][326][335]. Strategia aktywnego nadzoru pozwala leczyć jedynie klinicznie istotne przypadki raka gruczołu krokowego oraz jednocześnie unikać niepotrzebnego leczenia u chorych nieodnoszących korzyści z takiego leczenia[335].
Aktywny nadzór jest jedną z możliwych metod postępowania u wybranych chorych z potencjalnie uleczalnym rakiem o niskim ryzyku[325]. Kwalifikacja do aktywnego nadzoru odbywa się w oparciu o ustalone czynniki prognostyczne przebiegu miejscowo ograniczonego raka stercza, do których należy wiek, stopień zaawansowania, stopień złośliwości w skali Gleasona, rozległość zajęcia stercza oraz stężenie PSA[324]. Według Europejskiego Towarzystwa Urologicznego (EUA) aktywny nadzór oferuje się osobom z przewidywanym okresem przeżycia powyżej 10 lat z zaawansowaniem raka gruczołu krokowego pT1–pT2, stopniem złośliwości według klasyfikacji Gleasona nie wyższym niż 6, maksymalnie dwiema pozytywnymi biopsjami, naciekiem raka poniżej 50% objętości bioptatu oraz stężeniem PSA poniżej 10 ng/ml[336].
Monitorowanie chorych polega na okresowym badaniu lekarskim z badaniem przez odbyt, oceną stężenia PSA oraz ponowną biopsją gruczołu krokowego. Nie ustalono standardowego optymalnego schematu wykonywania badań kontrolnych w ramach aktywnego nadzoru i różne towarzystwa onkologiczne sugerują ich różną częstość[337][338]. Generalnie co 6 miesięcy chorego bada się przez odbyt oraz ocenia się stężenie wolnego oraz całkowitego PSA, a co 2–3 lata wykonuje się ponowną biopsję stercza[324]. EUA proponuje co 6 miesięcy ocenę stężenia PSA oraz badanie przez odbyt oraz okresowe ponowne biopsje stercza z przynajmniej 3–5 letnimi odstępami między nimi[339]. Decyzja o przejściu z nadzoru do leczenia może być oparta o progresję stopnia złośliwości w skali Gleasona, wzroście liczby biopsji (rdzeni) z rakiem lub wzroście stopnia ich zajęcia albo progresji stopnia zaawansowania miejscowego (cecha T według klasyfikacji TNM)[339].
Bezpieczeństwo i skuteczność aktywnego nadzoru oceniono w kilku badaniach klinicznych[340][326]. W dużym badaniu kohortowym obejmującym 993 mężczyzn z miejscowo ograniczonym rakiem gruczołu krokowego poddanych aktywnemu nadzorowi, w związku z progresją stężenia PSA, progresją według klasyfikacji Gleasona w kolejnych biopsjach lub preferencją chorego, ostatecznie 27% poddanych aktywnemu nadzorowi przeszło radykalne leczenie. Jedynie u 3% rozwinęły się przerzuty odległe. Odsetek 10-letniego przeżycia swoistego dla choroby wyniósł 98,1%, a odsetek 15-letniego przeżycia swoistego dla choroby 94,3%[341][325]. Z kolei w badaniu ProtecT 1643 mężczyzn z miejscowo ograniczonym rakiem gruczołu krokowego losowo przydzielono do grupy poddanej aktywnego nadzorowi, radykalnej prostatektomii lub radykalnej radioterapii i po 10 latach obserwacji nie stwierdzono istotnej różnicy mediany czasu przeżycia swoistego dla nowotworu[342][326]. Podobnie inne badania potwierdzają bezpieczeństwo aktywnego nadzoru, a opóźnienie rozpoczęcia leczenia radykalnego nie wpływa na wynik leczenia[343][344][345][346][347][326].
| Niskie ryzyko | Pośrednie ryzyko | Wysokie ryzyko |
| PSA < 10 ng/dl i | PSA 10–20 ng/dl lub | PSA > 20 ng/dl lub |
Ścisła obserwacja
Ścisła obserwacja (leczenie odroczone, leczenie objawowe) jest strategią zachowawczą dedykowaną chorym obciążonymi poważnymi chorobami współistniejącymi o przewidywanym przeżyciu poniżej 10 lat mającą na celu podtrzymanie możliwie wysokiej jakości życia i ograniczeniu toksyczności związanej z leczeniem. Ścisła obserwacja ma założenie paliatywne, czyli leczenie jest wdrażane wówczas, gdy progresja choroby jest przyczyną istotnych dolegliwości i również leczenie ma na celu podtrzymanie jakości życia (leczenie paliatywne). Założenie paliatywne odróżnia strategię ścisłej obserwacji od aktywnej obserwacji, która jest metodą racjonalizacji momentu wdrożenia leczenia o założeniu potencjalnie radykalnym u chorych z miejscowo ograniczonym rakiem o niskim ryzyku progresji i przewidywanym przeżyciu powyżej 10 lat w oparciu o obserwację historii naturalnej choroby i ocenę znanych czynników predykcyjnych jej przebiegu[325][348][334].
Prostatektomia



Radykalna prostatektomia jest operacją polegającą na wycięciu całego gruczołu krokowego wraz z odcinkiem sterczowym cewki moczowej oraz pęcherzykami nasiennymi. Operacja może być wykonywana z dostępu załonowego, a rzadziej dostępu kroczowego, metodą otwartą, laparoskopową albo z użyciem robota[349]. Celem operacji jest usunięcie całego nowotworu z wolnym od nacieku nowotworowego marginesem chirurgicznym[350]. Radykalna prostatektomia jest metodą leczenia zarezerwowaną dla chorych z miejscowo ograniczonym rakiem gruczołu krokowego w dobrym stanie sprawności bez poważnych schorzeń współwystępujących oraz przewidywanym czasie przeżycia powyżej 10 lat[349][350].
Prostatektomię z dostępu załonowego wykonuje się w znieczuleniu ogólnym, poprzez nacięcie w linii środkowej otwiera się przestrzeń zaotrzewnową. Na tym etapie wykonuje się limfadenektomię, jeśli jest ona konieczna. Po nacięciu powięzi ściennej miednicy uzyskuje się dostęp do wierzchołka gruczołu krokowego oraz błoniastej części cewki moczowej. Po zamknięciu żyły grzbietowej prącia na wysokości więzadła łukowatego łonowego oddziela się błoniastą część cewki moczowej od gruczołu krokowego. U podstawy stercza nacinana jest powięź trzewna miedniczna (powięź Denonvilliersa), co pozwala na dostęp i wycięcie pęcherzyków nasiennych oraz przecięcie nasieniowodów. W kolejnym etapie operacji przecina się szyję pęcherza moczowego i usuwa gruczoł krokowy. Ciągłość dróg moczowych jest odtwarzana poprzez zwężenie i zszycie szyi pęcherza moczowego z kikutem części błoniastej cewki moczowej. Zachowanie nerwów jamistych prącia wymaga odpowiedniej modyfikacji preparacji stercza pozwalającej zachować pęczki nerwowe przebiegające między odbytnicą a gruczołem krokowym[351][352]. Prostatektomia z dostępu kroczowego nie umożliwia wykonania limfadenektomii[353][349].
Elementem radykalnej prostatektomii może być usunięcie niektórych grup węzłów chłonnych nazywane limfadenektomią. Ze względu na wysoce zmienny obszar spływu chłonki z gruczołu krokowego nie ustalono optymalnego zakresu anatomicznego limfadenektomii[354]. Limfadenektomia może obejmować jedynie węzły biodrowe zewnętrzne lub węzły chłonne całego obszaru ograniczonego przez żyłę biodrową zewnętrzną od przodu, dno miednicy z tyłu, ścianę miednicy bocznie oraz ścianę pęcherza moczowego przyśrodkowo (rozszerzona limfadenektomia)[355][356]. Choć zajęcie węzłów chłonnych pogarsza rokowanie, to korzyść z wykonania limfadenektomii jest niepewna[350]. Kilka badań sugeruje poprawę przeżycia u chorych, u których wykonano rozszerzoną limfadenektomię[357][358][359][360], jednak późniejszy przegląd systematyczny nie wykazał żadnych korzyści w przeżyciu chorych[361]. Limfadenektomia dostarcza ważnych informacji dotyczących stadium choroby oraz rokowania, których nie można uzyskać inną metodą[362][356]. Nomogramy przedoperacyjne obejmujące różne czynniki prognostyczne takiej jak nomogram Briganti, MSKCC czy Roacha pomagają oszacować ryzyko zajęcia węzłów chłonnych i ustalić konieczność wykonania limfadenektomii[356][350]. Europejskie Towarzystwo Urologiczne sugeruje wykonywanie limfadenektomii, gdy ryzyko występowania przerzutów w węzłach chłonnych wynosi powyżej 5%[356]. Inni autorzy wskazują na inne wartości tego ryzyka[363][350].
Kilka badań wskazuje, że u mężczyzn z rakiem stercza o pośrednim oraz wysokim ryzyku radykalna prostatektomia ma przewagę nad ścisłą obserwacją, ponieważ u nich operacja pozwala zmniejszyć ryzyko rozwoju przerzutów oraz zgonu chorego. Natomiast u chorych z rakiem o niskim ryzyku badania dowodzą bezpieczeństwa aktywnego nadzoru i chorzy nie wymagają natychmiastowego leczenia radykalnego za pomocą prostatektomii czy radioterapii[364]. We wcześniej wspomnianym badaniu ProtecT u chorych z rakiem gruczołu krokowego o niskim ryzyku po 10 latach obserwacji nie wykazano przewagi radykalnej prostatektomii w zakresie poprawy przeżycia całkowitego chorych (OS) oraz przeżycia swoistego dla nowotworu (CSS)[342]. Podobnie w badaniu PIVOT po 10 latach obserwacji radykalna prostatektomia nie zmniejszyła śmiertelności z powodu raka[365][336]. Z kolei u chorych z rakiem o pośrednim ryzyku w badaniu SPCG-4 radykalna prostatektomia zmniejszała ryzyko zgonu oraz prawdopodobieństwo rozwoju przerzutów odległych[366]. W badaniu PIVOT u chorych z rakiem o pośrednim ryzyku zaobserwowano zmniejszenie śmiertelności z dowolnej przyczyny, jednak nie potwierdzono zmniejszenia śmiertelności z powodu raka gruczołu krokowego[365][367].
Chorzy z rakiem stercza o wysokim ryzyku mają wyższe ryzyko nawrotu, progresji z wytworzeniem przerzutów oraz zgonu z powodu choroby nowotworowej. U chorych w grupie wysokiego ryzyka, jeśli nie ma nacieku nowotworowego ściany miednicy lub zwieraczy cewki moczowej, zwykle wykonuje się prostatektomię z rozszerzoną limfadenektomią[367]. Również chorzy z rakiem o wysokim ryzyku odnoszą korzyść z wykonania prostatektomii[368]. Ocenia się, że 60% chorych z rakiem o wysokiej złośliwości (powyżej 8 stopni w skali Gleasona) po prostatektomii i dalszym leczeniu obejmującym hormonoterapię lub radioterapię osiąga piętnastoletnie przeżycie swoiste dla nowotworu[367]. W przypadku choroby zaawansowanej miejscowo w stadium cT3b-T4 prostatektomia w połączeniu z innymi metodami leczenia pozwala na osiągnięcie pięcioletniego przeżycia swoistego dla nowotworu u 87% leczonych[369][370][371][356].
Najważniejszymi niekorzystnymi następstwami operacji jest ryzyko zaburzeń wzwodu i nietrzymania moczu. Zaburzenia erekcji pojawiają się u ponad 50% leczonych, około 40–70% z nich jest leczona za pomocą inhibitorów PDE5 (sildenafil, tadalafil, wardenafil), inni potrzebują podania prostaglandyny E1, urządzenia próżniowego lub rzadziej protezy. Nietrzymanie moczu pojawia się u 5% poddanych operacji, jest związane z uszkodzeniem zewnętrznego zwieracza cewki moczowej. Leczenie polega na ćwiczeniach mięśni dna miednicy oraz wykorzystaniu technik biofeedbacku[372].
Leczenie uzupełniające po prostatektomii
U chorych w stadium zaawansowania miejscowego pT3pN0 o wysokim ryzyku wznowy miejscowej z powodu dodatniego marginesu chirurgicznego, przerwania torebki stercza, nacieku pęcherzyków nasiennych oraz ze stężeniem PSA poniżej 0,1 ng/ml stosuje się adiuwantową radioterapię[356].
Hormonoterapia w skojarzeniu z leczeniem chirurgicznym nie poprawia rokowania leczonych, ponieważ nie wpływa na zmniejszenie ryzyka wznowy miejscowej, wydłużenie mediany czasu przeżycia wolnego od progresji (PFS) oraz przeżycia całkowitego (OS)[373][374][356][375]. Jedynie u chorych po radykalnej prostatektomii z limfadenektomią z obecnymi przerzutami w węzłach chłonnych adiuwantowa ablacja androgenowa poprawia medianę przeżycia całkowitego (OS) i swoistego dla choroby nowotworowej (CSS)[376][377].
Inne metody leczenia miejscowego (terapia ogniskowa)
Nowe metody leczenia miejscowego raka gruczołu krokowego są badane jako alternatywa dla prostatektomii lub radioterapii w leczeniu pierwotnym miejscowo ograniczonego raka lub leczenia choroby nawrotowej[378]. Głównym celem metod ablacyjnych – krioterapii, HIFU, terapii fotodynamicznej, elektroporacji, CyberKnife – jest ogniskowe i selektywne leczenie choroby ograniczonej do gruczołu krokowego przy zaoszczędzeniu pęczków naczyniowo-nerwowych, zwieraczy cewki moczowej i cewki moczowej[379]. Dostępne wyniki badań nie pozwalają jednak uznać HIFU i krioterapii stercza za metody równoważne standardowym metodom terapii, jak leczenie chirurgiczne i radioterapia[380].
- Skupiona wiązka fal ultradźwiękowych o dużym natężeniu (HIFU)
Skupiona wiązka fal ultradźwiękowych o dużym natężeniu (ang. high-intensity focused ultrasound of the prostate, HIFU) jest metodą wykorzystującą ultradźwięki emitowane z przetwornika umieszczonego w odbytnicy w celu termicznego i mechanicznego uszkodzenia tkanki powodującej jej martwicę[381][382].
Nie przeprowadzono badań z obserwacją odległych wyników leczenia (powyżej 10 lat) tą metodą[383]. W przeglądzie systematycznym 20 badań klinicznych na około 4000 chorych poddanych HIFU w leczeniu pierwotnym choroby ograniczonej do gruczołu stwierdzono, że w przypadku zastosowania HIFU ryzyko nawrotu biochemicznego w ciągu roku było istotnie wyższe niż podczas stosowania radioterapii, jednak po 5 latach obserwacji różnica już nie była istotna statystycznie. Podobnie przeżycie wolne od choroby (DFS) po roku obserwacji było gorsze u stosujących HIFU niż u stosujących radioterapię, jednak po 3 latach obserwacji różnica również nie była istotna statystycznie[384][379].
Metodę HIFU oceniono w leczeniu choroby nawrotowej po radioterapii[385]. W prospektywnej analizie rejestru chorych z nawrotem po radioterapii leczonych za pomocą HIFU stwierdzono 63-miesięczną medianę przeżyć wolnych od nawrotu biochemicznego i 94% odsetek pięcioletnich przeżyć swoistych dla choroby nowotworowej[386][385].
- Krioterapia stercza
Krioterapia wykorzystuje techniki zamrażania w celu zniszczenia komórek przez odwodnienie komórki, denaturację białek, zniszczenie błon komórkowych oraz wywołanie zastoju w naczyniach w guzie z utworzeniem mikrozakrzepów powodujących niedokrwienie i martwicę. Zamrożenie tkanek gruczołu krokowego jest uzyskiwane przez wprowadzenie odpowiednich igieł przez odbytnicę pod kontrolą ultrasonografii przezodbytniczej, zwykle stosuje się kilka cykli zamrażania i rozmrażania. Potencjalnymi kandydatami do leczenia tą metodą są jedynie chorzy z rakiem o niskim lub pośrednim ryzyku, z nowotworem ograniczonym do stercza lub jedynie minimalnym szerzeniem się poza gruczoł, ze stężeniem PSA poniżej 20 ng/dl i objętością stercza poniżej 40 ml[383].
Dostępne są jedynie ograniczone dane dotyczące odległych wyników leczenia (powyżej 10 lat)[383]. W przeglądzie systematycznym porównującym krioterapię z prostatektomią oraz radioterapią oceniającym 19 badań klinicznych na blisko 4000 chorych stwierdzono, że roczne przeżycie wolne od choroby było gorsze u chorych stosujących krioterapię, a pozostałe parametry, w tym przeżycie swoiste dla choroby nowotworowej czy przeżycie całkowite, nie osiągnęły istotności statystycznej[384][383].
Radykalna radioterapia
Radioterapia jest metodą radykalnego leczenia miejscowo ograniczonego raka gruczołu krokowego[387]. W leczeniu mogą być wykorzystywane techniki teleradioterapii i brachyterapii[388]. Radioterapia jest stosowana samodzielnie lub w połączeniu z hormonoterapią[387].
Wybór metody radioterapii jest dostosowany do ustalonej grupy ryzyka chorego na raka stercza. U chorych z chorobą o niskim ryzyku preferuje się wykorzystanie brachyterapii lub ewentualnie stosuje się radioterapię z modulacją intensywności wiązki (IMRT)[389]. Chorzy z rakiem o pośrednim i wysokim ryzyku wymagają dawki naświetlania około 80 Gy. U chorych z rakiem o pośrednim lub wysokim ryzyku wykorzystuje się radioterapię z pól zewnętrznych (EBRT) stosowaną samodzielnie lub w skojarzeniu z brachyterapią o niskiej (LDR) lub wysokiej mocy dawki (HDR)[390][391]. Nie ma dostępnych wyników randomizowanych badań klinicznych porównujących różne techniki radioterapii[389].
Radioterapia z pól zewnętrznych

Radioterapia z pól zewnętrznych (EBRT) jest metodą radioterapii polegającą na podaniu promieniowania ze źródła położonego w pewnej odległości od leczonego. Metody planowania rozkładu dawki pozwoliły na precyzyjne określenie położenia gruczołu krokowego oraz otaczających struktur, co umożliwiło dopasowanie wiązki promieniowania do kształtu i objętości guza i ograniczenie objętości napromieniowywanych tkanek przy możliwości podania większej dawki promieniowania w obszarze nowotworu (radioterapia konformalna)[392][393]. W technice modulacji intensywności wiązki (IMRT) wykorzystuje się ruchome przesłony umieszczone w przebiegu wiązki w kolimatorze, które odpowiednio kierowane zgodnie z opracowanym planem ekspozycji optymalizują rozkład promieniowania i dopasowują je do gruczołu krokowego, przy jednoczesnym zaoszczędzeniu innych prawidłowych struktur[394][393]. Planowanie rozkładu dawki promieniowania jest wykonywane komputerowo w oparciu o badania obrazowe pomagające określić występujące stosunki anatomiczne[388][394]. Obecnie technika modulacji intensywności dawki jest standardową metodą radioterapii raka gruczołu krokowego[391][388].
Kliniczny obszar napromieniowania (CTV) obejmuje cały gruczoł krokowy oraz tkanki w najbliższym otoczeniu gruczołu krokowego, w tym pęcherzyki nasienne. U chorych o niskim ryzyku obszar napromieniowania może pominąć pęcherzyki nasienne[395]. Kilka badań wskazuje na poprawę czasu przeżycia wolnego od nawrotu w związku z eskalacją dawek z około 70 Gy do 80 Gy[396][397][398][399][400]. Korzyści odnoszą chorzy z rakiem o pośrednim i wysokim ryzyku, a nie odnoszą ich chorzy o niskim ryzyku[401][402]. W związku z tym najczęściej wykorzystuje się minimalną dawkę całkowitą przekraczającą 74 Gy[391][402].
Podanie większych pojedynczych dawek radioterapii (frakcji) bez zmiany dawki łącznej jest nazywane hipofrakcjonowaniem[403][404]. Frakcjonowanie radioterapii jest oparte na różnicy w możliwości naprawy materiału genetycznego w obrębie nowotworu i w prawidłowej tkance, która w szybko proliferujących komórkach jest niższa[405]. Zastosowanie wyższych pojedynczych dawek może sprzyjać nasileniu niszczenia komórek nowotworowych, a także skraca czas leczenia[404]. W raku stercza, ze względu na powolną proliferację, obserwuje się niski stosunek α/β wynoszący 1,5[403]. Sugeruje to wyższą wrażliwość na zwiększoną dawkę promieniowania podaną w jednej frakcji w porównaniu do konwencjonalnych frakcji (1,8–2 Gy)[403][406][405]. Większość badań oceniających zastosowanie hipofrakcjonowania to nierandomizowane serie z pojedynczych ośrodków na niewielkich grupach chorych[404]. W randomizowanych badaniach klinicznych umiarkowane hipofrakcjonowanie radioterapii z zastosowaniem 2,4–4 Gy na frakcję przez 4–6 tygodni wykazuje podobną lub nie niższą skuteczność od radioterapii frakcjonowanej konwencjonalnie[407][408][409][355]. Brakuje danych dotyczących długotrwałych obserwacji skuteczności hipofrakcjonowania leczenia[410].
U chorych z rakiem gruczołu krokowego o niskim ryzyku stosuje się radioterapię z pól zewnętrznych z modulacją intensywności dawki (IMRT) bez uzupełniającej hormonoterapii lub wykorzystuje się brachyterapię[411]. W tej grupie chorych w ramach teleterapii zwykle podaje się 74–78 Gy[390][412].
W grupie chorych z rakiem o pośrednim ryzyku stosuje się radioterapię z pól zewnętrznych z modulacją intensywności dawki połączoną z 4–6 miesięczną ablacją androgenową (ADT). U chorych nie mogących stosować hormonoterapii w leczeniu wykorzystuje się radioterapię z pól zewnętrznych z modulacją intensywności dawki do łącznej dawki 76–80 Gy lub teleradioterapię z modulacją intensywności dawki łączy się z brachyterapią[411].
U chorych o wysokim ryzyku ze względu na możliwość nawrotu poza obszarem poddanym leczeniu stosuje się leczenie skojarzone[411]. Radioterapię z modulacją intensywności dawki (IMRT) podaje się w zwiększonej dawce do 76–80 Gy albo radioterapię z modulacją intensywności dawki do łącznej dawki 46–58 Gy łączy się z brachyterapią o wysokiej mocy dawki (HDR) do łącznej dawki 80 Gy[391][390]. Jeśli jest to możliwe, obszar napromieniowania poszerza się o węzły chłonne miednicy[411]. W leczeniu uzupełniającym konieczna jest długotrwała hormonoterapia polegająca na ablacji androgenowej (ADT) przez okres 2–3 lat[390].
Brachyterapia

.jpg)

Brachyterapia jest metodą radioterapii, w której źródło promieniowania jest umieszczane w bardzo bliskim sąsiedztwie lub bezpośrednio w obrębie zmian nowotworowych[413]. Źródło promieniowania może być wszczepiane w obręb gruczołu krokowego na stałe lub czasowo, emituje ono promieniowanie o odpowiedniej energii oddziałujące jedynie w zakresie stercza, co pozwala na uniknięcie niepotrzebnego napromieniowywania pęcherza moczowego i odbytnicy[414]. U chorych z miejscowo ograniczonym rakiem gruczołu krokowego brachyterapia może być stosowana samodzielnie lub w skojarzeniu z radioterapią z pól zewnętrznych[415]. Samodzielną brachyterapię stosuje się u chorych z rakiem gruczołu krokowego o niskim ryzyku i u wybranych chorych z rakiem o pośrednim ryzyku z niewielką objętością nowotworu. U chorych z rakiem o pośrednim ryzyku brachyterapię łączy się z radioterapią z pól zewnętrznych (EBRT) i w tej grupie chorych do skojarzonej radioterapii może być dołączona hormonoterapia (ablacja androgenowa, ADT) przez okres 4–6 miesięcy. U chorych z rakiem o wysokim ryzyku brachyterapię łączy się z radioterapią z pól zewnętrznych (EBRT) i takie leczenie skojarzone może być łączone z hormonoterapią przez okres 2–3 lat. U chorych z dużą objętością gruczołu krokowego lub bardzo niewielką objętością stercza oraz u chorych z wcześniejszą TURP występuje większe ryzyko działań niepożądanych, dlatego w tej grupie czasem brachyterapię może poprzedzić ablacja androgenowa (leczenie neoadiuwantowe)[412].
- Brachyterapia o niskiej mocy dawki
W brachyterapii o niskiej mocy dawki (LDR) wykorzystuje się niewielkie implanty umieszczane na stałe w obrębie gruczołu krokowego[416]. Najczęściej wykorzystuje się radioaktywne izotopy jodu (125I) o okresie półtrwania wynoszącym 60 dni lub palladu (103Pd) z czasem połowicznego rozpadu wynoszącym 17 dni. Okres aktywności 125I wynosi około 10 miesięcy, a 103Pd około 3 miesięcy. Typowo przy stosowaniu 125I wykorzystuje się łączną dawkę 114 Gy, a dla 103Pd 125 Gy[415].
Implanty zawierające radioaktywny izotop są umieszczane za pomocą odpowiednich igieł wkłuwanych w okolicy kroczowej pod kontrolą ultrasonografii przezodbytniczej (TRUS). Zabieg może być wykonany dwuetapowo z wizualizacją gruczołu krokowego za pomocą ultrasonografii przezodbytniczej oraz zaplanowaniem toru przebiegu igły i położenia implantów w pierwszym etapie, a następnie w drugim etapie umieszczeniem implantów zgodnie z wcześniej ustalonym ich położeniem[417]. Obecnie już większość zabiegów jest wykonywana jednoetapowo pod kontrolą ultrasonografii przezodbytniczej w czasie rzeczywistym[418]. Zabieg jest wykonywany w znieczuleniu ogólnym, a w celu lepszej wizualizacji cewki moczowej cewnikuje się pęcherz moczowy. Po umieszczeniu w odbytnicy sondy ultrasonografu wizualizuje się warunki anatomiczne gruczołu krokowego. Przy użyciu odpowiedniego oprogramowania komputerowego powstaje wstępny plan toru przebiegu igły służącej do umieszczenia implantu z izotopem. Następnie za pomocą odpowiedniej prowadnicy poprzez krocze wprowadza się igłę do gruczołu krokowego. Po kontroli prawidłowego położenia igły poprzez jej wycofywanie w odpowiednich odstępach umieszcza się implanty z izotopem promieniotwórczym. W pierwszej kolejności wszczepiane są implanty z izotopem w obrębie obwodowych części gruczołu, a później w pozostałej centralnej części. Położenie implantów może być kontrolowane w czasie fluoroskopii[417]. Liczba koniecznych do wszczepienia implantów zawierających radioaktywny izotop jest obliczana na podstawie ustalonej aktywności zastosowanych implantów[417]. Inne techniki wykorzystują systemy planowania leczenia, oceny położenia implantów oraz śródzabiegową dozymetrię[418].
Samodzielna brachyterapia o niskiej mocy dawki może być stosowana u chorych z rakiem o niskim ryzyku z zaawansowaniem miejscowym cT1b–T2a, stopniu złośliwości w skali Gleasona nie wyższym niż 6 oraz stężeniem PSA poniżej 10 pg/ml. Objętość gruczołu krokowego nie powinna przekraczać 50 cm³, a objawy ze strony gruczołu krokowego nie powinny przekraczać 12 punktów w międzynarodowej skali punktowej objawów towarzyszących chorobom gruczołu krokowego (international prostate symptom score, IPSS)[416][414].
- Brachyterapia o wysokiej mocy dawki

W brachyterapii o wysokiej mocy dawki (HRT) stosuje się źródła promieniowania umieszczane czasowo w obrębie gruczołu krokowego i podaje się większe dawki promieniowania w porównaniu do brachyterapii o niskiej mocy dawki[419]. Zwykle wykorzystuje się radioaktywny 192Ir[389]. Brachyterapia o wysokiej mocy dawki ze względu na możliwość kontrolowania czasu ekspozycji i pozycji źródła promieniowania pomaga zoptymalizować podanie radioterapii i zwiększyć dokładność dawki, a także zmniejszyć ryzyko występowania obszarów o gorszej dystrybucji dawki (cold spots)[389][418][419]. Inną zaletą jest brak stałych implantów będącymi źródłami promieniowania[419].
Technika wykonania jest podobna do brachyterapii o niskiej mocy dawki. Zabieg jest wykonywany w znieczuleniu ogólnym, polega on na wprowadzeniu przez krocze po odpowiedniej prowadnicy pod kontrolą ultrasonografii przezodbytniczej 12–22 igieł do gruczołu krokowego oraz otaczających tkanek. Po zobrazowaniu warunków anatomicznych gruczołu krokowego powstaje plan leczenia obejmujący czas przebywania źródeł promieniowania w określonej pozycji i modyfikację położenia źródeł w zakresie prowadnicy. Dopiero po ustaleniu planu leczenia do prowadnicy wprowadzane jest źródło promieniowania, które po zakończeniu leczenia jest usuwane[419].
Brachyterapia o wysokiej mocy dawki może być podana w pojedynczej frakcji lub w kilku frakcjach. Często łączy się ją z radioterapią z pól zewnętrznych (EBRT)[416]. U chorych z miejscowo ograniczonym rakiem gruczołu krokowego o wysokim ryzyku lub rakiem miejscowo zaawansowanym skojarzenie radioterapii z pól zewnętrznych (40–50 Gy) z brachyterapią o wysokiej mocy dawki pozwala na eskalację dawki przy zmniejszeniu ryzyka toksyczności leczenia[420][421][422]. Skojarzenie radioterapii z zewnętrznych pól z brachyterapią o wysokiej mocy dawki poprawia wyniki końcowe leczenia, w tym czas przeżycia całkowitego (OS) i czas wolny od nawrotu (RFS)[423][424][425]. U chorych z wysokim ryzykiem nawrotu skojarzoną radioterapię EBRT i HDR zwykle łączy się z 2–3 letnią ablacją androgenową (hormonoterapia)[422]. U chorych z rakiem o niskim i wybranych chorych o pośrednim ryzyku radioterapia o wysokiej mocy dawki może być stosowana samodzielnie (bez EBRT)[416][426].
Hormonoterapia w leczeniu skojarzonym z radioterapią
Hormonoterapia jest elementem radykalnego leczenia skojarzonego z radioterapią, którego celem jest poprawa wyników leczenia[427]. Hormonoterapia może być leczeniem poprzedzającym, jednoczasowym lub uzupełniającym radioterapię[375]. Leczenie hormonalne raka gruczołu krokowego jest ukierunkowane na zależność tego nowotworu od androgenów. Polega na blokadzie wytwarzania androgenów w jądrach za pomocą agonistów gonadoliberyny (GnRH) lub antagonistów gonadoliberyny albo przez zablokowanie receptora androgenowego (AR) antyandrogenami (steroidowymi lub niesteroidowymi)[331].
W leczeniu skojarzonym z radioterapią hormonoterapia znajduje zastosowanie u chorych z rakiem o pośrednim i wysokim ryzyku. U chorych o pośrednim ryzyku ablacja androgenowa może być stosowana przez 4–6 miesięcy (tzw. krótka hormonoterapia), a u chorych z rakiem o wysokim ryzyku ablacja androgenowa jest podawana przez 2–3 lata[390]. W kilku dużych badaniach u chorych w rakiem gruczołu krokowego o wysokim ryzyku stosujących połączenie EBRT z ablacją androgenową (ADT) stwierdzono poprawę przeżycia swoistego dla choroby oraz przeżycia całkowitego w porównaniu z chorymi otrzymującymi wyłącznie samodzielną radioterapię[428][429][430][431][432][433][434]. Podobnie w kilku randomizowanych badaniach wykazano poprawę przeżycia całkowitego oraz przeżycia swoistego dla choroby nowotworowej u chorych o pośrednim ryzyku nawrotu po radioterapii stosujących 4–6 miesięczną ablację androgenową[435][436][437][438][439]. W badaniu RTOG 9910 u chorych poddanych radioterapii z rakiem o pośrednim ryzyku porównywano skuteczność ablacji androgenowej trwającej 4 miesiące z leczeniem trwającym 9 miesięcy i nie wykazano przewagi leczenia trwającego dłużej niż 4 miesiące[440]. W grupie chorych z rakiem gruczołu krokowego o niskim ryzyku stosuje się aktywny nadzór, prostatektomię lub radioterapię z pól zewnętrznych albo brachyterapię bez uzupełniającej hormonoterapii[327]. Samodzielna ablacja androgenowa, czyli bez połączenia z radioterapią, w grupie chorych z miejscowo ograniczonym (czyli bez przerzutów) rakiem gruczołu krokowego o niskim ryzyku nie poprawia przeżycia całkowitego leczonych i jest stosowana jedynie w leczeniu o założeniu paliatywnym[441][442][439].
Hormonoterapia paliatywna


Hormonoterapia jest podstawową metodą leczenia zaawansowanego raka gruczołu krokowego[330][443]. Rak gruczołu krokowego wykazuje zależność od androgenów, szczególnie od metabolitu testosteronu dihydrotestosteronu, dlatego ograniczenie dostępu androgenów dla komórek nowotworowych prowadzi do zahamowania wzrostu raka, pozwala opóźnić progresję choroby i łagodzi dolegliwości związane z chorobą nowotworową[443].
Opracowano dwie strategie hormonoterapii raka stercza. Farmakologiczne lub chirurgiczne zablokowanie lub ograniczenie produkcji testosteronu do poziomu kastracyjnego jest nazywane ablacją androgenową (androgen deprivation therapy, ADT)[443]. Produkcja androgenów jest kontrolowana przez oś przysadka-podwzgórze. Pulsacyjnie wydzielana przez podwzgórze gonadoliberyna (GnRH, LHRH) jest niezbędna do produkcji lutropiny (LH) stymulującej produkcję androgenów przez komórki Leydiga w jądrach oraz folikulotropiny (FSH) oddziałującej na komórki Sertolego, stymulując spermatogenezę[444]. Agonisty LHRH poprzez aktywację odpowiednich receptorów początkowo powodują wzrost stężenia LH i przejściowy wzrost stężenia androgenów. Jednak ciągła stymulacja receptorów LHRH w przysadce w ciągu 1–2 tygodni powoduje desensytyzację tych receptorów i regulację w dół ich ekspresji, ostatecznie osiąga się spadek stężenia LH i spadek stężenia androgenów do poziomu kastracyjnego[445][446][447][448]. Do analogów gonadoliberyny zalicza się goserelinę, leuprorelinę oraz tryptorelinę[391]. Jednak początkowy wzrost stężenia androgenów podczas rozpoczynania leczenia agonistami LHRH u części chorych z zaawansowanym nowotworem może doprowadzić do przejściowego zaostrzenia objawów choroby[445]. Mechanizm działania antagonistów LHRH jest oparty o kompetycyjną inhibicję receptora LHRH[449]. Antagonisty LHRH powodują zablokowanie produkcji LH i FSH, co skutkuje szybkim spadkiem stężenia androgenów bez okresu przejściowego wzrostu stężenia androgenów[448]. Do antagonistów LHRH zalicza się degareliks oraz abareliks[375]. Ryzyko reakcji anafilaktycznej, jaka towarzyszyła podawaniu abareliksu w badaniach klinicznych wpłynęło na niewielkie zainteresowanie lekiem i w 2005 roku producent zrezygnował z jego sprzedaży w Stanach Zjednoczonych. Natomiast degareliks (Firmagon®; Ferring Pharmaceuticals A/S) jest nowoczesnym antagonistą LHRH charakteryzującym się znacznie lepszym profilem bezpieczeństwa w porównaniu z abareliksem. Lek został zarejestrowany przez amerykańską Food and Drug Administration 24 grudnia 2008 roku do leczenia zaawansowanego raka stercza, a European Medicines Agency wydała zgodę na jego stosowanie w lutym 2009 roku[450].
W hormonoterapii raka stercza mogą być wykorzystywane steroidowe lub niesteroidowe antyandrogeny, które hamują oddziaływanie androgenów na komórki nowotworowe poprzez zablokowanie receptora androgenowego[443]. Niesteroidowe antyandrogeny, jak flutamid, nilutamid, bikalutamid czy enzalutamid, wykazują wysokie powinowactwo do receptora androgenowego, kompetycyjnie blokują go i uniemożliwiają transkrypcję genów zależnych od tego receptora. Steroidowe antyandrogeny, jak octan cyproteronu, poza kompetycyjną blokadą receptora androgenowego dodatkowo hamują uwalnianie gonadotropin i w tym mechanizmie obniżają stężenie androgenów[451]. Jednoczesne skojarzone zastosowanie ablacji androgenowej i antyandrogenu jest nazywane maksymalną blokadą androgenową (maximum androgen blockade, MAB)[375].
W przypadku raka gruczołu krokowego z obecnymi przerzutami odległymi w leczeniu stosuje się[452]:
- agonistę LHRH,
- agonistę LHRH z czasowym włączeniem antyandrogenu w celu redukcji ryzyka przejściowego zaostrzenia choroby (flare up),
- agonistę LHRH w skojarzeniu z antyandrogenem,
- ablację androgenową w skojarzeniu z chemioterapią z zastosowaniem 6 kursów docetakselu z lub bez prednizolonu.
U chorych z objawami klinicznymi choroby hormonoterapię włącza się po rozpoznaniu choroby[453]. Hormonoterapia u tych chorych przyczynia się do poprawy nasilenia objawów choroby oraz zmniejsza ryzyko poważnych powikłań choroby w tym zespołu ucisku rdzenia kręgowego, złamań patologicznych, niedrożności moczowodu czy przerzutów pozakostnych[453][454]. Mniej oczywisty jest optymalny moment włączenia ablacji androgenowej w bezobjawowej chorobie z przerzutami[455][456][457]. Według Europejskiego Towarzystwa Urologicznego wczesna ablacja androgenowa może być zastosowana w celu zapobiegnięcia progresji choroby bezobjawowej do choroby objawowej oraz wystąpienia poważnych powikłań. Kolejną opcją u bezobjawowych chorych z chorobą przerzutową jest odroczenie hormonoterapii do czasu progresji do choroby objawowej[453]. Badania porównujące skuteczność natychmiastowego włączenia hormonoterapii paliatywnej i odroczonej hormonoterapii paliatywnej zostały przeprowadzone przed wprowadzeniem oznaczeń PSA do oceny postępu choroby[457]. Stwierdzono w nich wyższość wczesnego wdrożenia ablacji androgenowej nad leczeniem odroczonym w zakresie poprawy przeżycia wolnego od progresji (PFS) oraz ryzyka wystąpienia powikłań związanych z chorobą, jednak nie zaobserwowano w nich istotnej statystycznie poprawy przeżycia całkowitego (OS) ani przeżycia swoistego dla choroby nowotworowej (CSS) związanego z wczesnym wdrożeniem ablacji androgenowej[458][457][459][460].
U większości chorych leczeniem z wyboru jest ablacja androgenowa (ADT) z zastosowaniem agonistów LHRH lub antagonistów LHRH[461][319]. Ablacja androgenowa u 60–70% leczonych powoduje spadek PSA do stężenia poniżej 4 ng/ml, u 30–50% chorych osiąga zmniejszenie mierzalnych zmian nowotworowych o przynajmniej połowę, ponad 60% chorych z objawami kostnych lub ze strony układu moczowego doświadcza złagodzenia dolegliwości[462]. Nie wykazano istotnej przewagi żadnej formy ablacji androgenowej, a także poszczególnych agonistów LHRH lub antagonistów LHRH[461][462][320][321]. Jedynie u chorych zagrożonych zespołem ucisku rdzenia kręgowego preferuje się obustronną orchidektomię lub antagonisty LHRH[461].
W pierwszej linii hormonoterapii nie stosuje się antyandrogenów w monoterapii[455]. Badania kliniczne porównujące antyandrogeny do chirurgicznej kastracji sugerują, że wykorzystanie antyandrogenów w pierwszej linii leczenia jest mniej skuteczne, ponieważ w mniejszym stopniu przyczyniają się do wydłużenia przeżycia całkowitego i ryzyka progresji[463][464].
Zastosowanie całkowitej blokady androgenowej (MAB), czyli połączenia ablacji androgenowej oraz antyandrogenu w pierwszej linii hormonoterapii wiąże się ze zwiększonym ryzykiem działań niepożądanych bez istotnego wpływu na poprawę przeżycia leczonych[454][464][455]. W badaniu Eisenbergera i współpracowników nie wykazano przewagi dołączenia flutamidu do obustronnej orchidektomii w porównaniu do samodzielnego zabiegu chirurgicznego[465][464]. Jednak w badaniach przeglądowych stwierdzono, że zastosowanie innych metod ablacji androgenowej prowadzi do niewielkiej korzyści w poprawie przeżycia całkowitego (poprawa odsetka przeżyć pięcioletnich poniżej 5%), choć wiąże się z istotnym zwiększeniem toksyczności leczenia[466][467][468][464]. Zastosowanie całkowitej blokady androgenowej może być przydatne jedynie w leczeniu chorych z dużą masą nowotworową, u których konieczne jest uzyskanie wysokiej odpowiedzi na leczenie[454].
U chorych z obecnymi przerzutami w momencie rozpoznania choroby w dobrym stanie sprawności, bez istotnych obciążeń chorobowych może być stosowane połączenie ablacji androgenowej z chemioterapią z użyciem docetakselu z prednizonem[453]. Kilka badań wskazuje, że połączenie docetakselu z ablacją androgenową w porównaniu do samodzielnej ablacji androgenowej wydłuża medianę przeżycia całkowitego[469][470][471][472][473].
- Przerywana hormonoterapia
U niektórych bezobjawowych chorych, u których za pomocą ablacji androgenowej uzyskano odpowiedź kliniczną i biochemiczną, może być oferowana przerywana hormonoterapia mająca na celu poprawę jakości życia i zmniejszenie działań niepożądanych leczenia[453][455]. Przerywana hormonoterapia polega na stosowaniu ablacji androgenowej do czasu uzyskania nadiru stężenia PSA i wówczas leczenie jest przerywane aż do czasu ponownego włączenia z powodu wzrostu stężenia PSA lub wystąpienia dolegliwości związanych z nowotworem[454]. Warunkiem wdrożenia takiej terapii jest uzyskanie wyraźnej odpowiedzi stężenia PSA ze spadkiem stężeniem markera do poziomu poniżej 4 ng/ml, a chory w trakcie przerwy w leczeniu jest poddawany okresowej kontroli lekarskiej[453][474]. Ponowną hormonoterapię wdraża się, gdy stężenie PSA wzrośnie do 10–20 ng/ml lub pojawią się objawy choroby[453].
W największym badaniu oceniającym bezpieczeństwo przerywanej hormonoterapii u chorych z chorobą z przerzutami nie osiągnięto wymaganego progu pozwalającego uznać, że leczenie przerywane nie jest gorsze od ciągłego i badanie nie pozwala wykluczyć wręcz gorszego przeżycia podczas leczenia przerywanego. W badaniu odnotowano niewielką korzyść w poprawie jakości życia leczonych przerywaną ablacją andogenową[475]. Z kolei w innych badaniach podczas leczenia przerywanego zaobserwowano przeżycie całkowite nie gorsze niż podczas leczenia ciągłego[476][477][464].
Większość przeprowadzonych badań była badaniami typu badana interwencja nie jest gorsza (ang. non-inferiority study)[478][464]. Tego typu badania polegają na porównaniu nowej metody do określonego standardu z przyjęciem progu istotności klinicznej, który pozwala interpretować, czy dana różnica skuteczności między dwoma ocenianymi grupami na niekorzyść badanej metody pozwala jeszcze określić ją jako nie gorszą od metody przyjętej jako wzorzec[479]. Nie wykazano przewagi w zakresie poprawy przeżycia leczonych metodą przerywanej ablacji androgenowej, a ponadto obserwuje się trend poprawy przeżycia całkowitego oraz przeżycia wolnego od progresji u chorych leczonych ciągłą ablacją androgenową[464].
Leczenie nawrotu biochemicznego
Najczęstszą formą nawrotu po leczeniu radykalnym jest wzrost stężenia PSA bez obecnej wykrywalnej choroby w badaniach obrazowych[480]. Choć u większości chorych po radykalnej prostatektomii czy radykalnej radioterapii uzyskuje się kontrolę choroby, to jednak około 20–40% leczonych prostatektomią oraz 30–50% leczonych radioterapią po 10 latach doświadcza nawrotu choroby[481]. Utrzymywanie się podwyższonego stężenia PSA lub wzrost stężenia PSA po początkowej normalizacji stężenia markera po leczeniu radykalnym może być związany z miejscowym nawrotem lub rozsiewem choroby. U części utrzymywanie się podwyższonego stężenia PSA jest związane z powolnym metabolizmem markera lub obecnością łagodnych chorób[482]. Istotne dla rokowania i dalszego leczenia jest wyróżnienie chorych z nawrotem miejscowym wymagających leczenia miejscowego od chorych z nawrotem z obecnymi przerzutami wymagających leczenia ogólnoustrojowego[482].
Diagnostyka u tych chorych zwykle obejmuje ocenę dynamiki wzrostu stężenia PSA, ponowną biopsję pod kontrolą TRUS, MRI gruczołu krokowego, a w przypadku pojawienia się objawów lub szybkiego czasu podwojenia stężenia PSA ocenę zajęcia kości. U części chorych może się okazać przydatna tomografia lub rezonans magnetyczny brzucha z miednicą oraz PET-TK z użyciem znakowanej choliny[482].
- Nawrót biochemiczny po radykalnej prostatektomii
W przypadku nawrotu pod postacią wzrostu stężenia PSA dalsze leczenie może obejmować[483]:
- radioterapię,
- ablację androgenową,
- obserwację.
Obserwacja jest opcją dla mężczyzn o stosunkowo długim czasie podwojenia stężenia PSA wynoszącym powyżej 12 miesięcy, nawrocie biochemicznym choroby powyżej 3 lat od leczenia radykalnego, stopniu złośliwości w skali Gleasona poniżej 7 oraz zaawansowaniu poniżej T3a albo oczekiwanym czasie życia poniżej 10 lat[484].
Wczesna radioterapia ratunkowa może dać możliwość wyleczenia chorych z nawrotem biochemicznym lub utrzymującym się podwyższonym stężeniem PSA po zabiegu[485]. Duże retrospektywe badania kohortowe wskazują na poprawę przeżycia swoistego dla choroby nowotworowej oraz zmniejszenie ryzyka zgonu[486][487][488]. Ratunkowa radioterapia może być łączona z ablacją androgenową, ponieważ poprawia ona wyniki leczenia[489][437][488].
Ablacja androgenowa w ramach leczenia nawrotu biochemicznego bywa wykorzystywana u chorych z wysokim podejrzeniem obecności przerzutów[488]. Podobnie jak w hormonoterapii choroby z przerzutami, w leczeniu ogólnoustrojowym u chorych z bezobjawowym nawrotem biochemicznym zwykle nie rozpoczyna się od razu ablacji androgenowej (ADT)[490]. Niektóre badania kliniczne sugerują korzyść ze wczesnej ablacji androgenowej[491], jednak inne badania nie wykazały różnicy między wczesną ablacją androgenową a odroczoną ablacją androgenową[492][493]. Jednak ze względu na niejasne korzyści stosowania hormonoterapii w leczeniu nawrotu biochemicznego, w tym niejasny wpływ na poprawę przeżycia leczonych oraz wpływ na jakość życia, nie wszyscy chorzy wymagają jej stosowania, ponieważ tylko u części chorych nawrót biochemiczny będzie ulegał dalszej progresji do choroby z przerzutami. Europejskie Towarzystwo Urologiczne proponuje stosowanie wczesnej ablacji androgenowej po nawrocie biochemicznym jedynie u chorych z wysokim ryzykiem progresji, w tym o czasie podwojenia stężenia PSA poniżej 6–12 miesięcy, rakiem ze stopniem złośliwości w skali Gleasona powyżej 7 oraz długim oczekiwanym czasie przeżycia[493].
- Nawrót biochemiczny po radykalnej radioterapii
W przypadku nawrotu po radioterapii radykalnej leczenie może obejmować[483]:
- ratunkową prostatektomię,
- HIFU,
- krioterapię,
- brachyterapię,
- ablację androgenową,
- obserwację.
Ratunkowa prostatektomia po biochemicznej progresji po radykalnej radioterapii daje największe możliwości lokalnej kontroli choroby[484]. Jednak ratunkowa prostatektomia, w porównaniu do pierwotnej radykalnej prostatektomii, niesie znacznie większe ryzyko zdarzeń niepożądanych[494][495][496]. W metaanalizie oceniono, że metoda pozwala na osiągnięcie 70–83% odsetka dziesięcioletniego przeżycia swoistego dla nowotworu (CSS) oraz 54–89% odsetek dziesięcioletnich przeżyć całkowitych (OS)[496]. Ratunkowa prostatektomia jest opcją u chorych z niewielkim obciążeniem chorób współistniejących, o wysokiej oczekiwanej długości życia. Stężenie PSA przed ratunkową prostatektomią nie powinno przekraczać 10 ng/ml, złośliwość nowotworu 7 według klasyfikacji Gleasona, nie mogą być obecne cechy zajęcia lokalnych węzłów chłonnych ani obecności przerzutów odległych[497].
W przypadku nawrotu po radykalnej radioterapii nie wykorzystuje się ratunkowej (ponownej) radioterapii z pól zewnętrznych (EBRT). Jednak u wybranych chorych z potwierdzonym histopatologicznie nawrotem może być zastosowana brachyterapia o niskiej (LDR) lub wysokiej mocy dawki (HDR)[498]. Dostępne są wyniki kilku serii badań oceniających skuteczność brachyterapii ratunkowej[499][500][501][502]. Prawdopodobnie brachyterapia o wysokiej mocy dawki wiąże się z mniejszą liczbą działań niepożądanych[498].
Ratunkowa krioablacja (SCAP) może być stosowana jako metoda alternatywna do ratunkowej prostatektomii. Jedynie kilka badań oceniało skuteczność tej metody[503]. Pięcioletnie odsetki przeżyć wolnych od nawrotów biochemicznych w jednym badaniu sięgały 50–70%[504], w innym blisko 50%[505]. W badaniu porównującym ratunkową prostatektomię z ratunkową krioablacją pięcioletnie odsetki przeżyć wolnych od nawrotu oraz przeżycie całkowite było wyraźnie lepsze u chorych po ratunkowej prostatektomii[506][503].
HIFU według EAU może być metodą alternatywną dla ablacji, jednak nie ma opublikowanych wyników badań na dużych grupach chorych oceniających skuteczność tej metody[507]. Obserwacja jest opcją u chorych, którzy nie chcą lub nie mogą podjąć leczenia drugiej linii[490]. Nie wykazano przewagi hormonoterapii w leczeniu nawrotu biochemicznego po radioterapii radykalnej nad samodzielną obserwacją[506][490].
Leczenie raka opornego na kastrację
Biochemiczna lub radiologiczna progresja choroby, mimo osiągnięcia kastracyjnego poziomu testosteronu (poniżej 50 ng/dl lub 1,7 nmol/l) podczas ablacji androgenowej, jest odzwierciedleniem rozwoju raka opornego na kastrację (CRPC)[508][509]. Rak oporny na kastrację stanowi pewne spektrum różnych obrazów klinicznych, może się prezentować jako bezobjawowy wzrost stężenia PSA, mimo stosowania ADT, ale również jako choroba z przerzutami powodująca znaczne dolegliwości[510]. Pod względem klinicznym wyróżnia się nieprzerzutowy rak oporny na kastrację obejmujący chorobę ograniczoną do gruczołu krokowego oraz przerzutowy rak oporny na kastrację, który obejmuje wzorzec z przerzutami do kości bez zmian w narządach wewnętrznych, wzorzec z zajęciem węzłów chłonnych z lub bez zajęcia kości i narządów wewnętrznych oraz wzorzec z zajęciem narządów wewnętrznych z lub bez zajęcia kości[508].
W leczeniu pierwszej linii stosuje się chemioterapię z użyciem docetakselu, abirateron, enzalutamid, sipuleucel-T lub radioaktywny rad (223Ra)[511]. Nie przeprowadzono badań klinicznych porównujących między sobą skuteczność leków wpływających na wydłużenie przeżycia chorych na raka gruczołu krokowego opornego na kastrację i nie ustalono optymalnej sekwencji stosowania leków wpływających na poprawę przeżycia chorych[512][513][514]. Wybór leczenia chorych z rakiem gruczołu krokowego opornym na kastrację zależy od stanu sprawności chorego, obecności objawów choroby, chorób współistniejących oraz rozległości zajęcia i położenia przerzutów[515][513]. Ze względu na niewielką toksyczność część autorów, mimo braku dowodów na wyższą aktywność nad taksanami, faworyzuje stosowanie abirateronu lub enzalutamidu w pierwszej linii leczenia[514].
U chorych z przerzutami do narządów wewnętrznych zwykle preferuje się docetaksel z prednizonem w rytmie co 3 tygodnie. Inną opcją leczniczą u tych chorych jest enzalutamid. Abirateron nie był oceniany w badaniach klinicznych u osób z objawową chorobą nie przyjmujących wcześniej docetakselu, przy czym według części autorów również może być on stosowany u osób nie kwalifikujących się do docetakselu[516].
W leczeniu bezobjawowych przerzutów bez zajęcia narządów wewnętrznych najczęściej stosuje się enzalutamid lub abirateron. Docetaksel jest wykorzystywany w objawowej chorobie przerzutowej, w chorobie bezobjawowej zwykle stosuje się inne opcje lecznicze, choć może być on konieczny u chorych z cechami szybkiej progresji lub z przerzutami do narządów wewnętrznych[516].
- Ablacja androgenowa
U mężczyzn z rakiem opornym na kastrację, mimo progresji choroby podczas ablacji androgenowej, nadal konieczne jest utrzymywanie kastracyjnego stężenia testosteronu[333][509][508]. W dwóch badaniach retrospektywnych u chorych z rakiem gruczołu krokowego opornego na kastrację u stosujących ADT zaobserwowano poprawę przeżycia całkowitego[517][518][509].
- Abirateron
Abirateron jest wybiórczym inhibitorem cytochromu P450 C17 (CYP17), będącego enzymem posiadającym aktywność 17α-hydroksylazy i C17,20-liazy. CYP17 jest niezbędny do biosyntezy androgenów w jądrach, nadnerczach i komórkach raka gruczołu krokowego. Abirateron hamuje biosyntezę androgenów na etapie przemian pregnenolonu i progesteronu. Dochodzi do zmniejszenia syntezy testosteronu i tym samym 5-α-dihydrotestosteronu indukującego proliferację komórek sterczowych. Abirateron zmniejsza stężenie testosteronu i innych androgenów w surowicy w większym stopniu, aniżeli analogi LHRH lub orchidektomia. Poprzez hamowanie CYP17 zwiększa wytwarzanie mineralokortykosteroidów w nadnerczach. Abirateron jest inhibitorem CYP2D6 i CYP2C8[519][520].
Abirateron jest stosowany w połączeniu z małą dawką glikokortykosteroidu takiego jak prednizon lub prednizolon w celu łagodzenia wynikającego z leczenia działania mineralokortykotropowego[521][516]. Leku nie powinno się przyjmować z jedzeniem[516].
Abirateron jest jedną z opcji terapeutycznych w leczeniu pierwszej linii raka gruczołu krokowego opornego na kastrację[515]. Skuteczność leku w leczeniu pierwszej linii u chorych z rakiem gruczołu krokowego opornym na kastrację nie leczonych wcześniej chemioterapią oceniono w badaniu III fazy COU-AA-302[522]. W tym badaniu chorzy losowo otrzymywali abirateron z prednizonem lub placebo z prednizonem i wykazano znaczącą poprawę mediany przeżycia wolnego od radiologicznej progresji (16,5 miesiąca w grupie leczonych abirateronem i 8,3 miesiąca w grupie leczonej placebo z prednizonem) oraz mediany przeżycia całkowitego (34,7 miesiąca w grupie leczonych abirateronem i 30,3 miesiąca w grupie leczonej placebo z prednizonem)[523][524][521].
W Polsce dopiero we wrześniu 2019 r. dopuszczono ten lek do leczenia w ramach programu NFZ opornego na kastrację RGK przed chemioterapią i to pod wieloma warunkami.
W maju 2018 r. FDA zatwierdziła nowy preparat octanu abirateronu (Yonsa) w połączeniu z metyloprednizolonem do leczenia mężczyzn z przerzutowym rakiem prostaty opornym na kastrację (mCRPC). Zatwierdzenie oparto na wynikach badania fazy II, w którym porównano preparat octanu abirateronu Yonsa z oryginalnym preparatem Zytiga, który został zatwierdzony w 2011 r. W badaniu znanym jako STAAR Yonsa wykazała równoważność terapeutyczną z preparatem Zytiga, a oba leki wykazywały podobne spadki PSA o ≥50%[525].
- Enzalutamid
Enzalutamid jest niesteroidowym antyandrogenem drugiej generacji wykazującym zdolność do zablokowania przekazywania sygnałów przez receptor androgenowy. Mechanizm działania enzalutamidu jest nieco odmienny niż niesteroidowych antyandrogenów pierwszej generacji (flutamid, nilutamid, bikalutamid)[451]. Lek kompetycyjnie blokuje wiązanie androgenów z receptorem androgenowym, uniemożliwia przemieszczenie aktywnego receptora androgenowego do jądra oraz blokuje wiązanie aktywnego receptora androgenowego z DNA[526][451].
Skuteczność enzalutamidu w leczeniu chorych z rakiem gruczołu krokowego nie poddawanych wcześniej chemioterapii określiło badanie PREVAIL, w którym lek porównano z placebo i u leczonych tym lekiem zaobserwowano poprawę mediany przeżycia całkowitego oraz przeżycia wolnego od progresji radiologicznej. Jednak u chorych z przerzutami do narządów wewnętrznych korzyść ze stosowania enzalutamidu w poprawie przeżycia całkowitego była ograniczona[527][509].
- Sipuleucel-T
Sipuleucel-T jest autologiczną immunoterapią komórkową opartą o pobrane od chorego komórki dendrytyczne laboratoryjnie eksponowane na zrekombinowane białko fuzyjne. W pierwszym etapie leczenia w procesie leukaferezy z krwi zbiera się komórki jednojądrzaste. Następnie po dalszej preparatyce, w celu uniknięcia reakcji immunosupresyjnych, komórki dendrytyczne eksponuje się poza organizmem leczonego na zrekombinowane białko fuzyjne. Eksponowany antygen jest zbudowany z wysoce swoistej dla gruczołu krokowego sterczowej fosfatazy kwaśnej oraz modulującego odpowiedź immunologiczną czynnika stymulującego tworzenie kolonii granulocytów i makrofagów (GM-CSF). Po podaniu choremu preparatu eksponowane na zrekombinowany antygen komórki dendrytyczne prezentują antygen i indukują rozwój odpowiedzi immunologicznej skierowanej przeciw komórkom nowotworowym z udziałem limfocytów cytotoksycznych[528][529][530].
Sipuleucel-T stosuje się w leczeniu bezobjawowych chorych lub minimalnie objawowych chorych na raka opornego na kastrację[531]. Sipuleucel-T jest zarezerwowany dla chorych o dobrym stanie sprawności (ECOG 0–1), oczekiwanym czasie przeżycia powyżej 6 miesięcy, bez przerzutów do wątroby[532]. W badaniu D9902B porównującym skuteczność sipuleucelu-T z placebo zaobserwowano poprawę mediany przeżycia całkowitego u leczonych sipuleucelem-T, u których wynosiła ona 25,8 miesiąca, podczas gdy w grupie otrzymującej placebo mediana przeżycia całkowitego wynosiła 21,7 miesiąca[533].
- Docetaksel
Docetaksel jest cytostatykiem należącym do grupy taksanów. Docetaksel w skojarzeniu z prednizonem w rytmie co 21 dni jest preferowanym programem chemioterapii u chorych z przerzutowym rakiem gruczołu krokowego opornym na kastrację[532].
W badaniu TAX327 porównującym docetaksel z prednizonem i mitoksantron z prednizonem, stanowiącym przez wiele lat standardowy cytostatyk w leczeniu opornego na kastrację raka gruczołu krokowego, wykazano przewagę docetakselu, u chorych stosujących docetaksel zaobserwowano dłuższą medianę przeżycia całkowitego wynoszącą 18,9 miesiąca, a u chorych leczonych mitoksantronem mediana ta wynosiła 16,5 miesiąca[534][535][536]. Podobnie w badaniu SWOG 9916 zaobserwowano dłuższe przeżycie chorych leczonych docetakselem niż mitoksantronem[537][536]. Nie wykazano przewagi programów wielolekowych zawierających docetaksel nad monoterapią z docetakselem[538]. U chorych, którzy nie są kandydatami do docetakselu lub nie tolerują leku może być stosowany kabazytaksel[532].
- Leczenie przerzutów do kości
Rak gruczołu krokowego wykazuje wysoką skłonność do lokalizowania przerzutów do kości, co wiąże się ze znacznymi dolegliwościami bólowymi i ryzykiem wystąpieniem powikłań kostnych (skeletal-related events, SRE) obejmujących złamania patologiczne, zespół ucisku rdzenia kręgowego czy konieczność operacji lub radioterapii[539][531]. Dodatkowym czynnikiem komplikującym jest utrata masy kostnej związanej z ADT oraz koniecznością stosowania glikokortykosteroidów w celu redukcji toksyczności taksanów[530].
Leczenie bolesnych zmian przerzutowych do kości obejmuje stosowanie leków przeciwbólowych takich jak niesteroidowe leki przeciwzapalne i opioidy[540]. Radioterapia z pól zewnętrznych wykazuje wysoką skuteczność w opanowywaniu dolegliwości bólowych ze strony bolesnych pojedynczych przerzutów do kości[541][542][543]. Większość chorych w ciągu 1–2 tygodni osiąga złagodzenie dolegliwości bólowych[543]. Patologiczne złamanie kręgosłupa czy zespół ucisku rdzenia kręgowego może wymagać interwencji neurochirurgicznej lub radioterapii, glikokortykosteroidy pomagają uniknąć dalszych uszkodzeń wtórnych do obrzęku tkanek miękkich[515][544].
W leczeniu objawowych przerzutów do kości może być zastosowany 223Ra. Jest to radiofarmaceutyk wbudowywany w hydroksyapatyt kości w miejscach o wysokim obrocie kostnym, emituje on głównie promieniowanie α, przez co ekspozycja sąsiednich tkanek jest bardzo niewielka[540][545]. Jest to jedyny lek ukierunkowany na kości, który poprawia przeżycie chorych[546]. U chorych na raka gruczołu krokowego opornego na kastrację z objawowymi przerzutami, ale bez przerzutów do narządów wewnętrznych, otrzymujących wcześniej docetaksel lub niekwalifikujących się do leczenia docetakselem porównano radioaktywny rad do placebo. Wykazano poprawę przeżycia całkowitego, zmniejszenie ryzyka powikłań kostnych oraz dolegliwości bólowych[547]. W kolejnych badaniach zaobserwowano również skuteczność radiofarmaceutyku u chorych nie otrzymujących wcześniej docetakselu[548][549][545].
W celu zmniejszenia ryzyka wystąpienia powikłań kostnych u chorych z przerzutami do kości w leczeniu stosuje się kwas zoledronowy (bisfosfonian) lub denosumab (inhibitor RANKL). Ponadto stosuje się suplementację witaminy D oraz wapnia[511]. W badaniu III fazy porównującym denosumab z kwasem zoledronowym zaobserwowano poprawę mediany przeżycia wolnego od przerzutów kostnych u chorych przyjmujących denosumab, przy czym stwierdzono podobne ryzyko zdarzeń kostnych (SRE) i nie potwierdzono poprawy przeżycia całkowitego (OS)[550][515].
- Leczenie drugiej linii
U chorych z progresją po leczeniu docetakselem w pierwszej linii leczenia opcjami leczniczymi w drugiej linii są[551][552]:
- abirateron z prednizonem (jeśli we wcześniejszej linii stosowano enzalutamid),
- 223Ra w przypadku choroby bez rozsiewu do narządów wewnętrznych z dominującymi przerzutami do kości,
- kabazytaksel,
- sipuleucel-T u chorych bezobjawowych lub z minimalnymi objawami,
- mitoksantron z prednizonem,
- wtórna hormonoterapia:
- antyandrogen,
- ketokonazol z lub bez hydrokortyzonu,
- kortykosteroid,
- dietylostylbestrol lub inny estrogen,
- najlepsza terapia wspomagająca (BSC).
Nie wypracowano konsensusu co do leczenia kolejnej linii po progresji na docetakselu, w tym sekwencji stosowania poszczególnych leków[552]. Wybór metody leczenia jest oparty o stan sprawności chorego, obecność chorób współistniejących, lokalizację przerzutów, wcześniejsze leczenie i profil toksyczności leków[511].
Kabazytaksel jest cytostatykiem należącym do grupy taksanów stosowanym u chorych z progresją po leczeniu docetakselem[546]. Skuteczność leku w drugiej linii leczenia wykazano w badaniu TROPIC, w którym kabazytaksel w porównaniu do mitoksantronu poprawiał medianę przeżycia całkowitego (OS), medianę przeżycia wolnego od progresji (PFS), a także wywoływał lepszą odpowiedź radiologiczną i biochemiczną[553]. W kolejnych badaniach nie potwierdzono wyższości kabazytakselu nad docetakselem w ramach leczenia I linii[554][546].
Skuteczność abirateronu w leczeniu po wcześniejszej terapii docetakselem oceniono w badaniu COU-AA-301, w którym u chorych przyjmujących abirateron zaobserwowano lepszą medianę przeżycia całkowitego w porównaniu do placebo[555][556][546]. Podobnie, w badaniu AFFIRM, wykazano przewagę enzalutamidu nad placebo[557]. U wybranych chorych bez ewidentnej progresji po wcześniejszym leczeniu z docetakselem może być korzystne ponowne wdrożenie chemioterapii z docetakselem[558].
U chorych z progresją po leczeniu abirateronem lub enzalutamidem w pierwszej linii leczenia opcjami leczniczymi w drugiej linii są[551][516]:
- docetaksel z prednizonem,
- abirateron z prednizonem (jeśli we wcześniejszej linii stosowano enzalutamid),
- enzalutamid (jeśli we wcześniejszej linii stosowano abirateron),
- 223Ra w przypadku choroby bez rozsiewu do narządów wewnętrznych z dominującymi przerzutami do kości,
- sipuleucel-T u chorych bezobjawowych lub z minimalnymi objawami,
- wtórna hormonoterapia:
- antyandrogen,
- ketokonazol z lub bez hydrokortyzonu,
- kortykosteroid,
- dietylostylbestrol lub inny estrogen,
- najlepsza terapia wspomagająca (BSC).
Nie ma dostępnych wyników badań porównujących skuteczność docetakselu z nowymi lekami hornonalnymi (abirateron, enzalutamid)[516][514].
Rokowanie
| Stadium kliniczne | Odsetek przeżycia pięcioletniego |
| Stadium miejscowe[a] | 100%[b] |
| Stadium uogólnione (rozsiane)[c] | 29,8%[d] |
Weterynaria
Rak gruczołu krokowego w weterynarii jest stosunkowo rzadką chorobą, dotyczącą przede wszystkim psów[561][562]. Najczęściej rozpoznaje się gruczolakoraka, a rzadziej raka płaskonabłonkowego, raka urotelialnego oraz raka niezróżnicowanego[562].
Nowotwór charakteryzuje się agresywnym przebiegiem, w około 80% przypadków jest rozpoznawany, gdy obecne są już przerzuty[563]. Choroba może objawiać się krwiomoczem, utykaniem tylnych kończyn, bolesnością, utratą apetytu i utratą masy[562][563]. Badanie przez odbyt pozwala wykazać obecność twardej asymetrycznej masy w obrębie stercza. Rozpoznanie jest stawiane na podstawie badania cytologicznego lub histopatologicznego materiału tkankowego uzyskanego podczas biopsji przezodbytniczej, przezkroczowej lub otwartej[563]. Nie jest znana metoda pozwalająca na wyleczenie, kastracja nie jest skuteczna. W leczeniu paliatywnym stosuje się cystostomię, umieszcza się stent w drogach moczowych oraz podaje leki przeciwbólowe. Możliwe jest wykorzystanie chemioterapii z podaniem mitoksantronu[563].
Historia

Pierwszy opis raka gruczołu krokowego podał John Adams w 1853 roku[564][565]. Choroba nie była rozpoznawana, dopóki nie powodowała dolegliwości z powodu zablokowania dróg moczowych albo przerzutów do kości lub narządów wewnętrznych[564]. Przed XX wiekiem chirurgiczne leczenie raka gruczołu krokowego polegające na usunięciu obturujących mas stercza przeprowadzano wyłącznie w celu łagodzenia niedrożności dróg moczowych i nie wypracowano żadnej techniki usunięcia gruczołu krokowego. Pierwszą prostatektomię z powodu raka w 1904 roku wykonał Hugh Hampton Young. Zastosowana metoda stała się standardem leczenia przez kolejne 40 lat. Zabieg był wykonywany głównie z zamiarem paliatywnym, dopiero później upowszechnił się jako metoda leczenia z zamiarem wyleczenia. W 1945 roku Terrence Millin wprowadził operacje z dostępu załonowego, która miała przewagę nad metodą z dostępu kroczowego. W latach 80. wprowadzono kolejne modyfikacje techniki operacyjnej zmniejszające krwawienie i ułatwiające zachowanie pęczków naczyniowo-nerwowych[564].
Pierwsze próby stosowania radioterapii w celu leczenia raka stercza datują się na początek XX wieku[564], w 1904 roku teleradioterapię wykorzystano w celu leczenia zaawansowanej choroby[566]. Radioterapia wiązała się ze znacznymi działaniami niepożądanymi, a ponadto wówczas stosowane techniki nie pozwalały na skuteczne leczenie głęboko położonych struktur jak gruczoł krokowy[566][564]. Wprowadzenie hormonoterapii w latach 40. XX wieku spowodowało zmniejszenie zainteresowania radioterapią. Jednak opracowane w latach 50. wysokoenergetyczne systemy radioterapii z zastosowaniem kobaltu umożliwiło penetrację promieniowania do głębszych poziomów. Pojawiły się doniesienia o wyleczeniu chorych za pomocą radioterapii[564]. Również w latach 50. zaczęto stosować metody brachyterapii polegające na umieszczeniu igieł z radioaktywnym złotem w obręb gruczołu krokowego i okoliczne tkanki[566]. Dzięki wprowadzeniu w latach 80. nowych technik umieszczania radioaktywnych implantów, które poprawiły bezpieczeństwo i skuteczność leczenia, zwiększyło się zainteresowanie brachyterapią[564]. Kolejne postępy teleradioterapii były związane z wprowadzaniem nowych technik dostarczania promieniowania i planowania dawki, które umożliwiło zastosowanie wyższej dawki leczenia przy zaoszczędzeniu sąsiednich tkanek[564].
W latach 90. XIX wieku zaobserwowano zmniejszenie objętości gruczołu krokowego po chirurgicznej kastracji, przez co zaczęła być ona stosowana w celu zmniejszenia objawów niedrożności dróg moczowych[566]. W 1941 roku Charles Huggins powiązał raka gruczołu krokowego z aktywnością hormonalną i zaobserwował poprawę po podaniu estrogenów[566][564]. Za swoje odkrycie w 1966 roku otrzymał Nagrodę Nobla[564]. Kolejne postępy w hormonoterapii raka stercza są związane z rozwojem metod blokowania produkcji androgenów oraz oddziaływania androgenów z ich receptorami. Kluczowym odkryciem było poznanie roli i budowy LHRH, które pozwoliło na opracowanie analogów tego neurohormonu[564]. Pod koniec lat 70. było już wiadome, że podanie analogów LHRH powoduje spadek stężenia androgenów, co powodowało poprawę dolegliwości chorych[567][568][564]. W 1977 roku Andrew Schally i Roger Guillemin za odkrycia dotyczące budowy i roli hormonów podwzgórzowych otrzymali Nagrodę Nobla[564]. W latach 70. opracowano pierwszy antyandrogen – cyprateton[569][564].
Badania dotyczące chemioterapii w raku gruczołu krokowego prowadzono od lat 50. W małych badaniach z lat 50. i 60. oceniano leki alkilujące. W 1975 roku w leczeniu ogólnoustrojowym raka opornego na kastrację wprowadzono program złożony z 5-fluorouracylu i cyklofosfamidu[570]. Następnie w latach 90. standardem leczenia przerzutowego raka stercza stał się mitoksantron z kortykosteroidem[571][572]. Następnie w badaniu TAX327 zaobserwowano przewagę docetakselu nad mitoksantronem[534].
Uwagi
- ↑ Stadium miejscowe jest definiowane jako ograniczenie procesu nowotworowego wyłącznie do narządu wyjściowego, czyli w tym przypadku do gruczołu krokowego[560].
- ↑ Populacja 18 wybranych obszarów w Stanach Zjednoczonych w latach 2007–2013; mężczyźni wszystkich ras i w każdym wieku, łącznie.
- ↑ Stadium zaawansowane choroby oznacza obecność procesu nowotworowego w odległych strukturach anatomicznych związaną z wytworzeniem przerzutów nowotworowych[560].
- ↑ Populacja 18 wybranych obszarów w Stanach Zjednoczonych w latach 2007–2013; mężczyźni wszystkich ras i w każdym wieku, łącznie.
Przypisy
- ↑ a b c d e f g h i J. Ferlay, I. Soerjomataram, R. Dikshit, S. Eser i inni. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. „Int J Cancer”. 136 (5), 2015. DOI: 10.1002/ijc.29210. PMID: 25220842.
- ↑ a b c d e f L.A. Torre, F. Bray, R.L. Siegel, J. Ferlay i inni. Global cancer statistics, 2012. „CA Cancer J Clin”. 65 (2), s. 87–108, 2015. DOI: 10.3322/caac.21262. PMID: 25651787.
- ↑ a b c d e f L.A. Torre, R.L. Siegel, E.M. Ward, A. Jemal. Global Cancer Incidence and Mortality Rates and Trends-An Update. „Cancer Epidemiol Biomarkers Prev”. 25 (1), s. 16–27, 2016. DOI: 10.1158/1055-9965.EPI-15-0578. PMID: 26667886.
- ↑ Mottet i in. 2017 ↓, s. 12.
- ↑ a b c d e f g h A.R. Patel, E.A. Klein. Risk factors for prostate cancer. „Nat Clin Pract Urol”. 6 (2), s. 87–95, 2009. DOI: 10.1038/ncpuro1290. PMID: 19198622.
- ↑ S. Loeb, M.A. Bjurlin, J. Nicholson, T.L. Tammela i inni. Overdiagnosis and overtreatment of prostate cancer. „Eur Urol”. 65 (6), s. 1046–1055, 2014. DOI: 10.1016/j.eururo.2013.12.062. PMID: 24439788.
- ↑ a b K.J. Bell, C. Del Mar, G. Wright, J. Dickinson i inni. Prevalence of incidental prostate cancer: A systematic review of autopsy studies. „Int J Cancer”. 137 (7), s. 1749–1757, 2015. DOI: 10.1002/ijc.29538. PMID: 25821151.
- ↑ M.M. Center, A. Jemal, J. Lortet-Tieulent, E. Ward i inni. International variation in prostate cancer incidence and mortality rates. „Eur Urol”. 61 (6), s. 1079–1092, 2012. DOI: 10.1016/j.eururo.2012.02.054. PMID: 22424666.
- ↑ a b Mottet i in. 2017 ↓, s. 12–13.
- ↑ a b c d e f g M.N. Bashir. Epidemiology of Prostate Cancer. „Asian Pac J Cancer Prev”. 16 (13), s. 5137–5141, 2015. PMID: 26225642.
- ↑ M. Daniyal, Z.A. Siddiqui, M. Akram, H.M. Asif i inni. Epidemiology, etiology, diagnosis and treatment of prostate cancer. „Asian Pac J Cancer Prev”. 15 (22), s. 9575–9578, 2014. PMID: 25520069.
- ↑ a b Krzakowski i in. 2015 ↓, s. 740.
- ↑ a b Jon Schiller: Prostate Cancer. CreateSpace, 2010, s. 21. ISBN 978-1-4536-8235-7.
- ↑ a b c d e f g h i j DeVita, Lawrence i Rosenberg 2015 ↓, s. 939.
- ↑ a b c d e Andrzej Szczeklik, Piotr Gajewski: Interna Szczeklika 2014. Kraków: Medycyna Praktyczna, 2014, s. 2216. ISBN 978-83-7430-405-4.
- ↑ Malcolm Mason, Leslie Moffat: Prostate Cancer. OUP Oxford, 2010, s. 16–17. ISBN 978-0-19-957393-6.
- ↑ a b c d e Krzakowski i in. 2015 ↓, s. 741.
- ↑ Vincent T. DeVita, Theodore S. Lawrence, Steven A. Rosenberg: DeVita, Hellman, and Rosenberg’s Cancer Principles & Practice of Oncology. Wolters Kluwer Health, 2015, s. 1401. ISBN 978-1-4511-9294-0.
- ↑ a b c d e f g h i j k l m n M.F. Leitzmann, S. Rohrmann. Risk factors for the onset of prostatic cancer: age, location, and behavioral correlates. „Clin Epidemiol”. 4, s. 1–11, 2012. DOI: 10.2147/CLEP.S16747. PMID: 22291478.
- ↑ a b c O.W. Brawley. Prostate cancer epidemiology in the United States. „World J Urol”. 30 (2), s. 195–200, Apr 2012. DOI: 10.1007/s00345-012-0824-2. PMID: 22476558.
- ↑ a b c Eric A. Klein, J. Stephen Jones: Management of Prostate Cancer. Springer Science & Business Media, 2012, s. 4–10. ISBN 978-1-60761-259-9.
- ↑ P.H. Gann. Risk factors for prostate cancer. „Rev Urol”. 4 Suppl 5, 2002. PMID: 16986064.
- ↑ a b c N.R. Perdana, C.A. Mochtar, R. Umbas, A.R. Hamid. The Risk Factors of Prostate Cancer and Its Prevention: A Literature Review. „Acta Med Indones”. 48 (3), s. 228–238, 2016. PMID: 27840359.
- ↑ Canadian Cancer Society: Risk factors for prostate cancer. [dostęp 2018-01-29]. [zarchiwizowane z tego adresu].
- ↑ American Cancer Society: Prostate Cancer Risk Factors. [dostęp 2018-01-29]. [zarchiwizowane z tego adresu].
- ↑ a b S. Punnen, M.R. Cooperberg. The epidemiology of high-risk prostate cancer. „Curr Opin Urol”. 23 (4), s. 331–336, 2013. DOI: 10.1097/MOU.0b013e328361d48e. PMID: 23619582.
- ↑ P. Lichtenstein, N.V. Holm, P.K. Verkasalo, A. Iliadou i inni. Environmental and heritable factors in the causation of cancer-analyses of cohorts of twins from Sweden, Denmark, and Finland. „N Engl J Med”. 343 (2), s. 78–85, 2000. DOI: 10.1056/NEJM200007133430201. PMID: 10891514.
- ↑ C. Frank, M. Fallah, J. Ji, J. Sundquist i inni. The population impact of familial cancer, a major cause of cancer. „Int J Cancer”. 134 (8), s. 1899–1906, 2014. DOI: 10.1002/ijc.28510. PMID: 24590453.
- ↑ a b A. Brandt, J.L. Bermejo, J. Sundquist, K. Hemminki. Age-specific risk of incident prostate cancer and risk of death from prostate cancer defined by the number of affected family members. „Eur Urol”. 58 (2), s. 275–280, 2010. DOI: 10.1016/j.eururo.2010.02.002. PMID: 20171779.
- ↑ K. Hemminki. Familial risk and familial survival in prostate cancer. „World J Urol”. 30 (2), s. 143–148, 2012. DOI: 10.1007/s00345-011-0801-1. PMID: 22116601.
- ↑ K. Hemminki, J. Sundquist, J.L. Bermejo. How common is familial cancer?. „Ann Oncol”. 19 (1), s. 163–167, 2008. DOI: 10.1093/annonc/mdm414. PMID: 17804474.
- ↑ I.J. Powell. The precise role of ethnicity and family history on aggressive prostate cancer: a review analysis. „Arch Esp Urol”. 64 (8), s. 711–719, 2011. PMID: 22052754.
- ↑ L.E. Johns, R.S. Houlston. A systematic review and meta-analysis of familial prostate cancer risk. „BJU Int”. 91 (9), s. 789–794, 2003. PMID: 12780833.
- ↑ S.R. Potter, A.W. Partin. Hereditary and familial prostate cancer: biologic aggressiveness and recurrence. „Rev Urol”. 2 (1), s. 35–36, 2000. PMID: 16985733.
- ↑ G. Colloca, A. Venturino. The evolving role of familial history for prostate cancer. „Acta Oncol”. 50 (1), s. 14–24, 2011. DOI: 10.3109/0284186X.2010.521191. PMID: 20874046.
- ↑ a b C. Alberti. Hereditary/familial versus sporadic prostate cancer: few indisputable genetic differences and many similar clinicopathological features. „Eur Rev Med Pharmacol Sci”. 14 (1), s. 31–41, 2010. PMID: 20184087.
- ↑ a b Marta Heise, Olga Haus. Dziedziczny rak gruczołu krokowego. „Postepy Hig Med Dosw”, 2014.
- ↑ a b c O. Bratt. Hereditary prostate cancer: clinical aspects. „J Urol”. 168 (3), s. 906–913, 2002. DOI: 10.1097/01.ju.0000024402.67529.ca. PMID: 12187189.
- ↑ O. Bratt. Hereditary prostate cancer. „BJU Int”. 85 (5), s. 588–598, 2000. PMID: 10735934.
- ↑ B.S. Carter, T.H. Beaty, G.D. Steinberg, B. Childs i inni. Mendelian inheritance of familial prostate cancer. „Proc Natl Acad Sci U S A”. 89 (8), s. 3367–3371, 1992. PMID: 1565627.
- ↑ a b Cezary Cybulski, Bartosz Gliniewicz, Andrzej Sikorski, Jan Lubiński. Genetyka kliniczna raka prostaty. „Postępy Nauk Medycznych”, 2008.
- ↑ L.Q. Qin, J, Y. Xu, P, Y. Wang, J. Tong i inni. Milk consumption is a risk factor for prostate cancer in Western countries: evidence from cohort studies. „Asia Pac J Clin Nutr”. 16 (3), s. 467–476, 2007. PMID: 17704029.
- ↑ Y. Song, J.E. Chavarro, Y. Cao, W. Qiu i inni. Whole milk intake is associated with prostate cancer-specific mortality among U.S. male physicians. „J Nutr”. 143 (2), s. 189–196, 2013. DOI: 10.3945/jn.112.168484. PMID: 23256145.
- ↑ X. Gao, M.P. LaValley, K.L. Tucker. Prospective studies of dairy product and calcium intakes and prostate cancer risk: a meta-analysis. „J Natl Cancer Inst”. 97 (23), s. 1768–1777, 2005. DOI: 10.1093/jnci/dji402. PMID: 16333032.
- ↑ J.M. Chan, E. Giovannucci, S.O. Andersson, J. Yuen i inni. Dairy products, calcium, phosphorous, vitamin D, and risk of prostate cancer (Sweden). „Cancer Causes Control”. 9 (6), s. 559–566, 1998. PMID: 10189041.
- ↑ Y. Park, P.N. Mitrou, V. Kipnis, A. Hollenbeck i inni. Calcium, dairy foods, and risk of incident and fatal prostate cancer: the NIH-AARP Diet and Health Study. „Am J Epidemiol”. 166 (11), s. 1270–1279, 2007. DOI: 10.1093/aje/kwm268. PMID: 18000020.
- ↑ a b c A. Wolk. Diet, lifestyle and risk of prostate cancer. „Acta Oncol”. 44 (3), s. 277–281, 2005. DOI: 10.1080/02841860510029572. PMID: 16076700.
- ↑ D.D. Alexander, P.J. Mink, C.A. Cushing, B. Sceurman. A review and meta-analysis of prospective studies of red and processed meat intake and prostate cancer. „Nutr J”. 9. s. 50. DOI: 10.1186/1475-2891-9-50. PMID: 21044319.
- ↑ L.C. Bylsma, D.D. Alexander. A review and meta-analysis of prospective studies of red and processed meat, meat cooking methods, heme iron, heterocyclic amines and prostate cancer. „Nutr J”. 14, s. 125, 2015. DOI: 10.1186/s12937-015-0111-3. PMID: 26689289.
- ↑ D.D. Alexander, J.K. Bassett, D.L. Weed, E.C. Barrett i inni. Meta-Analysis of Long-Chain Omega-3 Polyunsaturated Fatty Acids (LCω-3PUFA) and Prostate Cancer. „Nutr Cancer”. 67 (4), s. 543–554, 2015. DOI: 10.1080/01635581.2015.1015745. PMID: 25826711.
- ↑ a b c Mottet i in. 2017 ↓, s. 14.
- ↑ a b c d E.H. Allott, E.M. Masko, S.J. Freedland. Obesity and prostate cancer: weighing the evidence. „Eur Urol”. 63 (5), s. 800–809, May 2013. DOI: 10.1016/j.eururo.2012.11.013. PMID: 23219374.
- ↑ A.G. Renehan, M. Tyson, M. Egger, R.F. Heller i inni. Body-mass index and incidence of cancer: a systematic review and meta-analysis of prospective observational studies. „Lancet”. 371 (9612), s. 569–578, 2008. DOI: 10.1016/S0140-6736(08)60269-X. PMID: 18280327.
- ↑ R.J. MacInnis, D.R. English. Body size and composition and prostate cancer risk: systematic review and meta-regression analysis. „Cancer Causes Control”. 17 (8), s. 989–1003, 2006. DOI: 10.1007/s10552-006-0049-z. PMID: 16933050.
- ↑ A. Bergström, P. Pisani, V. Tenet, A. Wolk i inni. Overweight as an avoidable cause of cancer in Europe. „Int J Cancer”. 91 (3), s. 421–430, 2001. PMID: 11169969.
- ↑ Y. Cao, J. Ma. Body mass index, prostate cancer-specific mortality, and biochemical recurrence: a systematic review and meta-analysis. „Cancer Prev Res (Phila)”. 4 (4), s. 486–501, 2011. DOI: 10.1158/1940-6207.CAPR-10-0229. PMID: 21233290.
- ↑ a b L.K. Dennis, D.V. Dawson. Meta-analysis of measures of sexual activity and prostate cancer. „Epidemiology”. 13 (1), s. 72–79, 2002. PMID: 11805589.
- ↑ W.Q. Lian, F. Luo, X.L. Song, Y.J. Lu i inni. Gonorrhea and Prostate Cancer Incidence: An Updated Meta-Analysis of 21 Epidemiologic Studies. „Med Sci Monit”. 21, s. 1902–1910, 2015. DOI: 10.12659/MSM.893579. PMID: 26126881.
- ↑ M.L. Taylor, A.G. Mainous, B.J. Wells. Prostate cancer and sexually transmitted diseases: a meta-analysis. „Fam Med”. 37 (7). s. 506–512. PMID: 15988645.
- ↑ S. Caini, S. Gandini, M. Dudas, V. Bremer i inni. Sexually transmitted infections and prostate cancer risk: a systematic review and meta-analysis. „Cancer Epidemiol”. 38 (4), s. 329–338, 2014. DOI: 10.1016/j.canep.2014.06.002. PMID: 24986642.
- ↑ S. Sutcliffe, E. Giovannucci, A.M. De Marzo, M.F. Leitzmann i inni. Gonorrhea, syphilis, clinical prostatitis, and the risk of prostate cancer. „Cancer Epidemiol Biomarkers Prev”. 15 (11), s. 2160–2166, 2006. DOI: 10.1158/1055-9965.EPI-05-0913. PMID: 17119041.
- ↑ W.Y. Huang, R. Hayes, R. Pfeiffer, R.P. Viscidi i inni. Sexually transmissible infections and prostate cancer risk. „Cancer Epidemiol Biomarkers Prev”. 17 (9), s. 2374–2381, 2008. DOI: 10.1158/1055-9965.EPI-08-0173. PMID: 18768506.
- ↑ A.F. Kotb, A. Beltagy, A.M. Ismail, M.M. Hashad. Sexual activity and the risk of prostate cancer: Review article. „Arch Ital Urol Androl”. 87 (3), s. 214–215, 2015. DOI: 10.4081/aiua.2015.3.214. PMID: 26428643.
- ↑ J.R. Rider, K.M. Wilson, J.A. Sinnott, R.S. Kelly i inni. Ejaculation Frequency and Risk of Prostate Cancer: Updated Results with an Additional Decade of Follow-up. „Eur Urol”. 70 (6), s. 974–982, 2016. DOI: 10.1016/j.eururo.2016.03.027. PMID: 27033442.
- ↑ M.F. Leitzmann, E.A. Platz, M.J. Stampfer, W.C. Willett i inni. Ejaculation frequency and subsequent risk of prostate cancer. „JAMA”. 291 (13), s. 1578–1586, 2004. DOI: 10.1001/jama.291.13.1578. PMID: 15069045.
- ↑ R.J. Shephard. Physical Activity and Prostate Cancer: An Updated Review. „Sports Med”. 47 (6), s. 1055–1073, 2017. DOI: 10.1007/s40279-016-0648-0. PMID: 27844337.
- ↑ C.M. Friedenreich, I. Thune. A review of physical activity and prostate cancer risk. „Cancer Causes Control”. 12 (5), s. 461–475, 2001. PMID: 11545461.
- ↑ a b c C. De Nunzio, G.L. Andriole, I.M. Thompson, S.J. Freedland. Smoking and Prostate Cancer: A Systematic Review. „Eur Urol Focus”. 1 (1), s. 28–38, 2015. DOI: 10.1016/j.euf.2014.10.002. PMID: 28723351.
- ↑ M. Huncharek, K.S. Haddock, R. Reid, B. Kupelnick. Smoking as a risk factor for prostate cancer: a meta-analysis of 24 prospective cohort studies. „Am J Public Health”. 100 (4), s. 693–701, 2010. DOI: 10.2105/AJPH.2008.150508. PMID: 19608952.
- ↑ F. Islami, D.M. Moreira, P. Boffetta, S.J. Freedland. A systematic review and meta-analysis of tobacco use and prostate cancer mortality and incidence in prospective cohort studies. „Eur Urol”. 66 (6), s. 1054–1064, 2014. DOI: 10.1016/j.eururo.2014.08.059. PMID: 25242554.
- ↑ P.H. Gann, C.H. Hennekens, J. Ma, C. Longcope i inni. Prospective study of sex hormone levels and risk of prostate cancer. „J Natl Cancer Inst”. 88 (16), s. 1118–1126, 1996. PMID: 8757191.
- ↑ N.E. Eaton, G.K. Reeves, P.N. Appleby, T.J. Key. Endogenous sex hormones and prostate cancer: a quantitative review of prospective studies. „Br J Cancer”. 80 (7), s. 930–944, 1999. DOI: 10.1038/sj.bjc.6690445. PMID: 10362098.
- ↑ A.W. Roddam, N.E. Allen, P. Appleby, T.J. Key i inni. Endogenous sex hormones and prostate cancer: a collaborative analysis of 18 prospective studies. „J Natl Cancer Inst”. 100 (3), s. 170–183, 2008. DOI: 10.1093/jnci/djm323. PMID: 18230794.
- ↑ M.A. Rowlands, D. Gunnell, R. Harris, L.J. Vatten i inni. Circulating insulin-like growth factor peptides and prostate cancer risk: a systematic review and meta-analysis. „Int J Cancer”. 124 (10), s. 2416–2429, 2009. DOI: 10.1002/ijc.24202. PMID: 19142965.
- ↑ A.G. Renehan, M. Zwahlen, C. Minder, S.T. O’Dwyer i inni. Insulin-like growth factor (IGF)-I, IGF binding protein-3, and cancer risk: systematic review and meta-regression analysis. „Lancet”. 363 (9418), s. 1346–1353, 2004. DOI: 10.1016/S0140-6736(04)16044-3. PMID: 15110491.
- ↑ J.K. Morris, L.M. George, T. Wu, N.J. Wald. Insulin-like growth factors and cancer: no role in screening. Evidence from the BUPA study and meta-analysis of prospective epidemiological studies. „Br J Cancer”. 95 (1), s. 112–117, 2006. DOI: 10.1038/sj.bjc.6603200. PMID: 16804529.
- ↑ A.W. Roddam, N.E. Allen, P. Appleby, T.J. Key i inni. Insulin-like growth factors, their binding proteins, and prostate cancer risk: analysis of individual patient data from 12 prospective studies. „Ann Intern Med”. 149 (7), s. 461–471, 2008. PMID: 18838726.
- ↑ a b G. Doolan, G. Benke, G. Giles. An update on occupation and prostate cancer. „Asian Pac J Cancer Prev”. 15 (2), s. 501–516, 2014. PMID: 24568454.
- ↑ T.J. Key. Fruit and vegetables and cancer risk. „Br J Cancer”. 104 (1), s. 6–11, 2011. DOI: 10.1038/sj.bjc.6606032. PMID: 21119663.
- ↑ R. Takachi, M. Inoue, N. Sawada, M. Iwasaki i inni. Fruits and vegetables in relation to prostate cancer in Japanese men: the Japan Public Health Center-Based Prospective Study. „Nutr Cancer”. 62 (1), s. 30–39, 2010. DOI: 10.1080/01635580903191502. PMID: 20043257.
- ↑ a b V.A. Kirsh, U. Peters, S.T. Mayne, A.F. Subar i inni. Prospective study of fruit and vegetable intake and risk of prostate cancer. „J Natl Cancer Inst”. 99 (15), s. 1200–1209, 2007. DOI: 10.1093/jnci/djm065. PMID: 17652276.
- ↑ P. Chen, W. Zhang, X. Wang, K. Zhao i inni. Lycopene and Risk of Prostate Cancer: A Systematic Review and Meta-Analysis. „Medicine (Baltimore)”. 94 (33), 2015. DOI: 10.1097/MD.0000000000001260. PMID: 26287411.
- ↑ D. Ilic, M. Misso. Lycopene for the prevention and treatment of benign prostatic hyperplasia and prostate cancer: a systematic review. „Maturitas”. 72 (4), s. 269–276, 2012. DOI: 10.1016/j.maturitas.2012.04.014. PMID: 22633187.
- ↑ E. Giovannucci, E.B. Rimm, Y. Liu, M.J. Stampfer i inni. A prospective study of cruciferous vegetables and prostate cancer. „Cancer Epidemiol Biomarkers Prev”. 12 (12), s. 1403–1409, 2003. PMID: 14693729.
- ↑ L. Yan, E.L. Spitznagel. Soy consumption and prostate cancer risk in men: a revisit of a meta-analysis. „Am J Clin Nutr”. 89 (4), s. 1155–1163, 2009. DOI: 10.3945/ajcn.2008.27029. PMID: 19211820.
- ↑ a b S.M. Lippman, E.A. Klein, P.J. Goodman, M.S. Lucia i inni. Effect of selenium and vitamin E on risk of prostate cancer and other cancers: the Selenium and Vitamin E Cancer Prevention Trial (SELECT). „JAMA”. 301 (1), s. 39–51, 2009. DOI: 10.1001/jama.2008.864. PMID: 19066370.
- ↑ G. Trottier, N. Lawrentschuk, N.E. Fleshner. Prevention strategies in prostate cancer. „Curr Oncol”. 17 Suppl 2, 2010. PMID: 20882132.
- ↑ a b c d Mottet i in. 2017 ↓, s. 17.
- ↑ a b c N. Bell, S. Connor Gorber, A. Shane, M. Joffres i inni. Recommendations on screening for prostate cancer with the prostate-specific antigen test. „CMAJ”. 186 (16), s. 1225–1234, 2014. DOI: 10.1503/cmaj.140703. PMID: 25349003.
- ↑ a b c K. Kohestani, M. Chilov, S.V. Carlsson. Prostate cancer screening-when to start and how to screen?. „Transl Androl Urol”. 7 (1), s. 34–45, 2018. DOI: 10.21037/tau.2017.12.25. PMID: 29594018.
- ↑ a b c Ashutosh Tewari: Prostate Cancer: A Comprehensive Perspective. Springer Science & Business Media, 2013, s. 347. ISBN 978-1-4471-2864-9.
- ↑ a b c d J.L. Silberstein, J.K. Parsons. Prostate cancer prevention: concepts and clinical recommendations. „Prostate Cancer Prostatic Dis”. 13 (4), s. 300–306, 2010. DOI: 10.1038/pcan.2010.18. PMID: 20567257.
- ↑ a b MayoClinic: Prostate cancer prevention: Ways to reduce your risk. [dostęp 2018-02-02].
- ↑ a b Mottet i in. 2017 ↓, s. 13.
- ↑ M.J. Barry, L.H. Simmons. Prevention of Prostate Cancer Morbidity and Mortality: Primary Prevention and Early Detection. „Med Clin North Am”. 101 (4), s. 787–806, 2017. DOI: 10.1016/j.mcna.2017.03.009. PMID: 28577627.
- ↑ Michael P. Pignone, Kirsten Bibbins-Domingo: Disease Prevention, An Issue of Medical Clinics of North America. Elsevier Health Sciences, 2017. ISBN 978-0-323-53139-9.
- ↑ National Cancer Institute: Prostate Cancer Prevention (PDQ®)–Patient Version. [dostęp 2018-02-02].
- ↑ a b H.K. Moorthy, P. Venugopal. Strategies for prostate cancer prevention: Review of the literature. „Indian J Urol”. 24 (3), s. 295–302, Jul 2008. DOI: 10.4103/0970-1591.42608. PMID: 19468457.
- ↑ a b Z. Hamilton, J.K. Parsons. Prostate Cancer Prevention: Concepts and Clinical Trials. „Curr Urol Rep”. 17 (4), s. 35, 2016. DOI: 10.1007/s11934-016-0587-1. PMID: 26957512.
- ↑ I.M. Thompson, P.J. Goodman, C.M. Tangen, M.S. Lucia i inni. The influence of finasteride on the development of prostate cancer. „N Engl J Med”. 349 (3), s. 215–224, 2003. DOI: 10.1056/NEJMoa030660. PMID: 12824459.
- ↑ a b c d I. Thompson, A. Kristal, E.A. Platz. Prevention of prostate cancer: outcomes of clinical trials and future opportunities. „Am Soc Clin Oncol Educ Book”, s. 76–80, 2014. DOI: 10.14694/EdBook_AM.2014.34.e76. PMID: 24857150.
- ↑ M.W. Redman, C.M. Tangen, P.J. Goodman, M.S. Lucia i inni. Finasteride does not increase the risk of high-grade prostate cancer: a bias-adjusted modeling approach. „Cancer Prev Res (Phila)”. 1 (3), s. 174–181, 2008. DOI: 10.1158/1940-6207.CAPR-08-0092. PMID: 19138953.
- ↑ I.M. Thompson, P.J. Goodman, C.M. Tangen, H.L. Parnes i inni. Long-term survival of participants in the prostate cancer prevention trial. „N Engl J Med”. 369 (7), s. 603–610, 2013. DOI: 10.1056/NEJMoa1215932. PMID: 23944298.
- ↑ L.C. Clark, G.F. Combs, B.W. Turnbull, E.H. Slate i inni. Effects of selenium supplementation for cancer prevention in patients with carcinoma of the skin. A randomized controlled trial. Nutritional Prevention of Cancer Study Group. „JAMA”. 276 (24), s. 1957–1963, 1996. PMID: 8971064.
- ↑ A.R. Kristal, A.K. Darke, J.S. Morris, C.M. Tangen i inni. Baseline selenium status and effects of selenium and vitamin e supplementation on prostate cancer risk. „J Natl Cancer Inst”. 106 (3), 2014. DOI: 10.1093/jnci/djt456. PMID: 24563519.
- ↑ a b Andrzej Stelmach, Łukasz Wohadlo. Badania przesiewowe w kierunku wykrywania raka stercza. Aktualny stan wiedzy i argumenty za skriningiem. „Nowotwory”, 2013.
- ↑ a b Eric A. Klein, J. Stephen Jones: Management of Prostate Cancer. Springer Science & Business Media, 2012, s. 26. ISBN 978-1-60761-259-9.
- ↑ Eric A. Klein, J. Stephen Jones: Management of Prostate Cancer. Springer Science & Business Media, 2012, s. 25–26. ISBN 978-1-60761-259-9.
- ↑ a b c d Mottet i in. 2017 ↓, s. 17–18.
- ↑ a b c d Mottet i in. 2017 ↓, s. 16.
- ↑ R. Etzioni, R. Gulati, M.R. Cooperberg, D.M. Penson i inni. Limitations of basing screening policies on screening trials: The US Preventive Services Task Force and Prostate Cancer Screening. „Med Care”. 51 (4), s. 295–300, 2013. DOI: 10.1097/MLR.0b013e31827da979. PMID: 23269114.
- ↑ a b c DeVita, Lawrence i Rosenberg 2015 ↓, s. 938.
- ↑ a b F.H. Schröder, J. Hugosson, M.J. Roobol, T.L. Tammela i inni. Screening and prostate-cancer mortality in a randomized European study. „N Engl J Med”. 360 (13), s. 1320–1328, 2009. DOI: 10.1056/NEJMoa0810084. PMID: 19297566.
- ↑ F.H. Schröder, J. Hugosson, M.J. Roobol, T.L. Tammela i inni. Prostate-cancer mortality at 11 years of follow-up. „N Engl J Med”. 366 (11), s. 981–990, 2012. DOI: 10.1056/NEJMoa1113135. PMID: 22417251.
- ↑ P.R. Carroll, J.K. Parsons. NCCN clinical practice guidelines in oncology: prostate cancer early detection. Version 1.2018 – March 12, 2018. „J Natl Compr Canc Netw”, s. 14–15, 2018.
- ↑ a b c P.R. Carroll, J.K. Parsons. NCCN clinical practice guidelines in oncology: prostate cancer early detection. Version 1.2018 – March 12, 2018. „J Natl Compr Canc Netw”, s. 15–16, 2018.
- ↑ J. Hugosson, S. Carlsson, G. Aus, S. Bergdahl i inni. Mortality results from the Göteborg randomised population-based prostate-cancer screening trial. „Lancet Oncol”. 11 (8), s. 725–732, 2010. DOI: 10.1016/S1470-2045(10)70146-7. PMID: 20598634.
- ↑ R. Arnsrud Godtman, E. Holmberg, H. Lilja, J. Stranne i inni. Opportunistic testing versus organized prostate-specific antigen screening: outcome after 18 years in the Göteborg randomized population-based prostate cancer screening trial. „Eur Urol”. 68 (3), s. 354–360, 2015. DOI: 10.1016/j.eururo.2014.12.006. PMID: 25556937.
- ↑ G.L. Andriole, E.D. Crawford, R.L. Grubb, S.S. Buys i inni. Mortality results from a randomized prostate-cancer screening trial. „N Engl J Med”. 360 (13), s. 1310–1319, 2009. DOI: 10.1056/NEJMoa0810696. PMID: 19297565.
- ↑ G.L. Andriole, E.D. Crawford, R.L. Grubb, S.S. Buys i inni. Prostate cancer screening in the randomized Prostate, Lung, Colorectal, and Ovarian Cancer Screening Trial: mortality results after 13 years of follow-up. „J Natl Cancer Inst”. 104 (2), s. 125–132, 2012. DOI: 10.1093/jnci/djr500. PMID: 22228146.
- ↑ P.F. Pinsky, P.C. Prorok, K. Yu, B.S. Kramer i inni. Extended mortality results for prostate cancer screening in the PLCO trial with median follow-up of 15 years. „Cancer”. 123 (4), s. 592–599, 2017. DOI: 10.1002/cncr.30474. PMID: 27911486.
- ↑ a b P.R. Carroll, J.K. Parsons. NCCN clinical practice guidelines in oncology: prostate cancer early detection. Version 1.2018 – March 12, 2018. „J Natl Compr Canc Netw”, s. 16, 2018.
- ↑ E.D. Crawford, R. Grubb, A. Black, G.L. Andriole i inni. Comorbidity and mortality results from a randomized prostate cancer screening trial. „J Clin Oncol”. 29 (4), s. 355–361, 2011. DOI: 10.1200/JCO.2010.30.5979. PMID: 21041707.
- ↑ D. Ilic, M.M. Neuberger, M. Djulbegovic, P. Dahm. Screening for prostate cancer. „Cochrane Database Syst Rev”, 2013. DOI: 10.1002/14651858.CD004720.pub3. PMID: 23440794.
- ↑ J.H. Hayes, M.J. Barry. Screening for prostate cancer with the prostate-specific antigen test: a review of current evidence. „JAMA”. 311 (11), s. 1143–1149, 2014. DOI: 10.1001/jama.2014.2085. PMID: 24643604.
- ↑ E.A. Heijnsdijk, E.M. Wever, A. Auvinen, J. Hugosson i inni. Quality-of-life effects of prostate-specific antigen screening. „N Engl J Med”. 367 (7), s. 595–605, 2012. DOI: 10.1056/NEJMoa1201637. PMID: 22894572.
- ↑ H. Vasarainen, H. Malmi, L. Määttänen, M. Ruutu i inni. Effects of prostate cancer screening on health-related quality of life: results of the Finnish arm of the European randomized screening trial (ERSPC). „Acta Oncol”. 52 (8), s. 1615–1621, 2013. DOI: 10.3109/0284186X.2013.802837. PMID: 23786174.
- ↑ N. Booth, P. Rissanen, T.L. Tammela, L. Määttänen i inni. Health-related quality of life in the Finnish trial of screening for prostate cancer. „Eur Urol”. 65 (1), s. 39–47, 2014. DOI: 10.1016/j.eururo.2012.11.041. PMID: 23265387.
- ↑ a b c d e f g J. Hugosson. Stopping screening, when and how?. „Transl Androl Urol”. 7 (1), s. 46–53, 2018. DOI: 10.21037/tau.2017.12.39. PMID: 29594019.
- ↑ Jakub Dobruch, Piotr L. Chłosta, Andrzej Borówka. Badania przesiewowe mające na celu wczesne wykrycie raka stercza w XXI wieku. „Postępy Nauk Medycznych”, 2012.
- ↑ a b DeVita, Lawrence i Rosenberg 2015 ↓, s. 937.
- ↑ a b C.H. Bangma, S. Roemeling, F.H. Schröder. Overdiagnosis and overtreatment of early detected prostate cancer. „World J Urol”. 25 (1), s. 3–9, 2007. DOI: 10.1007/s00345-007-0145-z. PMID: 17364211.
- ↑ K. Zhang, C.H. Bangma, M.J. Roobol. Prostate cancer screening in Europe and Asia. „Asian J Urol”. 4 (2), s. 86–95, 2017. DOI: 10.1016/j.ajur.2016.08.010. PMID: 29264211.
- ↑ a b c Ashutosh Tewari: Prostate Cancer: A Comprehensive Perspective. Springer Science & Business Media, 2013, s. 348. ISBN 978-1-4471-2864-9.
- ↑ a b G.S. Sandhu, G.L. Andriole. Overdiagnosis of prostate cancer. „J Natl Cancer Inst Monogr”. 2012 (45), s. 146–151, 2012. DOI: 10.1093/jncimonographs/lgs031. PMID: 23271765.
- ↑ a b c d e Mottet i in. 2017 ↓, s. 18.
- ↑ P.R. Carroll, J.K. Parsons. NCCN clinical practice guidelines in oncology: prostate cancer early detection. Version 1.2018 – March 12, 2018. „J Natl Compr Canc Netw”, s. 17, 2018.
- ↑ a b P.R. Carroll, J.K. Parsons. NCCN clinical practice guidelines in oncology: prostate cancer early detection. Version 1.2018 – March 12, 2018. „J Natl Compr Canc Netw”, s. 18–19, 2018.
- ↑ P.R. Carroll, J.K. Parsons. NCCN clinical practice guidelines in oncology: prostate cancer early detection. Version 1.2018 – March 12, 2018. „J Natl Compr Canc Netw”, s. 17–18, 2018.
- ↑ a b Christopher Sweeney: Prostate Cancer, An Issue of Hematology/Oncology Clinics of North America. Elsevier Health Sciences, 2013, s. 1096–1097. ISBN 978-0-323-22723-0.
- ↑ A. Auvinen. Prostate cancer screening: what can we learn from randomised trials?. „Transl Androl Urol”. 7 (1), s. 12–17, 2018. DOI: 10.21037/tau.2017.12.13. PMID: 29594015.
- ↑ a b P.R. Carroll, J.K. Parsons. NCCN clinical practice guidelines in oncology: prostate cancer early detection. Version 1.2018 – March 12, 2018. „J Natl Compr Canc Netw”, s. 19–20, 2018.
- ↑ K.S. Louie, A. Seigneurin, P. Cathcart, P. Sasieni. Do prostate cancer risk models improve the predictive accuracy of PSA screening? A meta-analysis. „Ann Oncol”. 26 (5), s. 848–864, 2015. DOI: 10.1093/annonc/mdu525. PMID: 25403590.
- ↑ J.M. Chan, E.L. Van Blarigan, S.A. Kenfield. What should we tell prostate cancer patients about (secondary) prevention?. „Curr Opin Urol”. 24 (3), s. 318–323, 2014. DOI: 10.1097/MOU.0000000000000049. PMID: 24625429.
- ↑ a b Fletcher 2013 ↓, s. 879.
- ↑ a b c d e f DeVita, Lawrence i Rosenberg 2015 ↓, s. 935.
- ↑ a b c d Krzakowski i in. 2015 ↓, s. 742.
- ↑ Eble i in. 2004 ↓, s. 160.
- ↑ Fletcher 2013 ↓, s. 873.
- ↑ Eble i in. 2004 ↓, s. 165.
- ↑ a b c Stachura i Domagała 2009 ↓, s. 998.
- ↑ Eble i in. 2004 ↓, s. 164.
- ↑ a b c d e f g h i Eble i in. 2004 ↓, s. 169.
- ↑ Eble i in. 2004 ↓, s. 169–170.
- ↑ a b c d Eble i in. 2004 ↓, s. 170.
- ↑ Stachura i Domagała 2009 ↓, s. 999.
- ↑ a b c d Eble i in. 2004 ↓, s. 171.
- ↑ a b c d Noel Weidner, Richard J. Cote, Saul Suster, Lawrence M. Weiss: Modern Surgical Pathology. Elsevier Health Sciences, 2009, s. 1143–1147. ISBN 978-1-4377-1958-1.
- ↑ Eble i in. 2004 ↓, s. 171–172.
- ↑ a b Eble i in. 2004 ↓, s. 199.
- ↑ a b Eble i in. 2004 ↓, s. 200.
- ↑ Eble i in. 2004 ↓, s. 200–201.
- ↑ a b c d e Eble i in. 2004 ↓, s. 202.
- ↑ a b c d e f g h i j k P.A. Humphrey. Histological variants of prostatic carcinoma and their significance. „Histopathology”. 60 (1), s. 59–74, 2012. DOI: 10.1111/j.1365-2559.2011.04039.x. PMID: 22212078.
- ↑ Eble i in. 2004 ↓, s. 205.
- ↑ Fletcher 2013 ↓, s. 906.
- ↑ a b c Eble i in. 2004 ↓, s. 206.
- ↑ a b Noel Weidner, Richard J. Cote, Saul Suster, Lawrence M. Weiss: Modern Surgical Pathology. Elsevier Health Sciences, 2009, s. 1149–1150. ISBN 978-1-4377-1958-1.
- ↑ Eble i in. 2004 ↓, s. 207.
- ↑ Eble i in. 2004 ↓, s. 208.
- ↑ Eble i in. 2004 ↓, s. 207–208.
- ↑ a b c d Eble i in. 2004 ↓, s. 179.
- ↑ a b c d e Noel Weidner, Richard J. Cote, Saul Suster, Lawrence M. Weiss: Modern Surgical Pathology. Elsevier Health Sciences, 2009, s. 1147–1148. ISBN 978-1-4377-1958-1.
- ↑ Stachura i Domagała 2009 ↓, s. 999–1000.
- ↑ a b Stachura i Domagała 2009 ↓, s. 1000.
- ↑ a b c d e f g h i j k l Eble i in. 2004 ↓, s. 180.
- ↑ a b c d e Michelle S. Hirsch: Genitourinary Pathology: An Issue of Surgical Pathology Clinics. Elsevier Health Sciences, 2016, s. 539–542. ISBN 978-0-323-39587-8.
- ↑ a b Richard J. Ablin, Malcolm D. Mason: Metastasis of Prostate Cancer. Springer Science & Business Media, 2007, s. 5–18. ISBN 978-1-4020-5847-9.
- ↑ M. Popiolek, J.R. Rider, O. Andrén, S.O. Andersson i inni. Natural history of early, localized prostate cancer: a final report from three decades of follow-up. „Eur Urol”. 63 (3), s. 428–435, 2013. DOI: 10.1016/j.eururo.2012.10.002. PMID: 23084329.
- ↑ J. Ramon, Denis Denis: Prostate Cancer. Springer Science & Business Media, 2007, s. 34. ISBN 978-3-540-40901-4.
- ↑ Ashutosh K. Tewari, Peter Whelan, John D. Graham: Prostate Cancer: Diagnosis and Clinical Management. John Wiley & Sons, 2013, s. 7–8. ISBN 978-1-118-34739-3.
- ↑ M. Thompson, Martin I. Resnick, Eric A. Klein: Prostate Cancer Screening. Springer Science, 2001. ISBN 978-1-4757-6306-5.
- ↑ M.E. Cowen, M. Chartrand, W.F. Weitzel. A Markov model of the natural history of prostate cancer. „J Clin Epidemiol”. 47 (1), s. 3–21, 1994. PMID: 8283192.
- ↑ a b c d e Rutkowski i Warzocha 2013 ↓, s. 336.
- ↑ a b c d e Stachura i Domagała 2009 ↓, s. 1001.
- ↑ a b c d Vincent T. Devita, Samuel Hellman, Steven A. Rosenberg: Cancer: Principles and Practice of Oncology 6th edition. Lippincott Williams & Wilkins Publishers, 2001, s. 1022–1023.
- ↑ a b c d e f g h B.S. Knudsen, V. Vasioukhin. Mechanisms of prostate cancer initiation and progression. „Adv Cancer Res”. 109, s. 1–50, 2010. DOI: 10.1016/B978-0-12-380890-5.00001-6. PMID: 21070913.
- ↑ a b William B. Coleman, Gregory J. Tsongalis: Molecular Pathology: The Molecular Basis of Human Disease. Academic Press, 2009, s. 489–495. ISBN 978-0-08-092219-5.
- ↑ a b DeVita, Lawrence i Rosenberg 2015 ↓, s. 936.
- ↑ Jeanne Held-Warmkessel: Contemporary Issues in Prostate Cancer: A Nursing Perspective. Jones & Bartlett Learning, 2009. ISBN 978-0-7637-3075-8.
- ↑ a b c L.N. Fonseka, M.E. Kallen, A. Serrato-Guillen, R. Chow i inni. Cytogenetics and Molecular Genetics of Prostate Cancer: A Comprehensive Update. „J Assoc Genet Technol”. 41 (3), s. 100–111, 2015. PMID: 26606177.
- ↑ M.M. Shen, C. Abate-Shen. Molecular genetics of prostate cancer: new prospects for old challenges. „Genes Dev”. 24 (18), s. 1967–2000, 2010. DOI: 10.1101/gad.1965810. PMID: 20844012.
- ↑ T. Mitchell, D.E. Neal. The genomic evolution of human prostate cancer. „Br J Cancer”. 113 (2), s. 193–198, 2015. DOI: 10.1038/bjc.2015.234. PMID: 26125442.
- ↑ A. Abeshouse, J. Ahn, R. Akbani, A. Ally i inni. The Molecular Taxonomy of Primary Prostate Cancer. „Cell”. 163 (4), s. 1011–1025, 2015. DOI: 10.1016/j.cell.2015.10.025. PMID: 26544944.
- ↑ a b c d e f g h i j S.D. Kaffenberger, C.E. Barbieri. Molecular subtyping of prostate cancer. „Curr Opin Urol”. 26 (3), s. 213–218, 2016. DOI: 10.1097/MOU.0000000000000285. PMID: 26986650.
- ↑ a b c DeVita, Lawrence i Rosenberg 2015 ↓, s. 928.
- ↑ a b R.L. Shand, E.P. Gelmann. Molecular biology of prostate-cancer pathogenesis. „Curr Opin Urol”. 16 (3), s. 123–131, 2006. DOI: 10.1097/01.mou.0000193384.39351.64. PMID: 16679847.
- ↑ a b D. Gasi Tandefelt, J. Boormans, K. Hermans, J. Trapman. ETS fusion genes in prostate cancer. „Endocr Relat Cancer”. 21 (3), 2014. DOI: 10.1530/ERC-13-0390. PMID: 24659477.
- ↑ a b c d e f g DeVita, Lawrence i Rosenberg 2015 ↓, s. 927.
- ↑ C. Kumar-Sinha, S.A. Tomlins, A.M. Chinnaiyan. Recurrent gene fusions in prostate cancer. „Nat Rev Cancer”. 8 (7), s. 497–511, 2008. DOI: 10.1038/nrc2402. PMID: 18563191.
- ↑ a b c d e f g h i M.H. Tan, J. Li, H.E. Xu, K. Melcher i inni. Androgen receptor: structure, role in prostate cancer and drug discovery. „Acta Pharmacol Sin”. 36 (1), s. 3–23, 2015. DOI: 10.1038/aps.2014.18. PMID: 24909511.
- ↑ Z. Culig, F.R. Santer. Androgen receptor signaling in prostate cancer. „Cancer Metastasis Rev”. 33 (2–3), s. 413–427, 2014. DOI: 10.1007/s10555-013-9474-0. PMID: 24384911.
- ↑ E. Jernberg, A. Bergh, P. Wikström. Clinical relevance of androgen receptor alterations in prostate cancer. „Endocr Connect”. 6 (8), 2017. DOI: 10.1530/EC-17-0118. PMID: 29030409.
- ↑ P.E. Lonergan, D.J. Tindall. Androgen receptor signaling in prostate cancer development and progression. „J Carcinog”. 10, s. 20, 2011. DOI: 10.4103/1477-3163.83937. PMID: 21886458.
- ↑ A.A. Shafi, A.E. Yen, N.L. Weigel. Androgen receptors in hormone-dependent and castration-resistant prostate cancer. „Pharmacol Ther”. 140 (3), s. 223–238, 2013. DOI: 10.1016/j.pharmthera.2013.07.003. PMID: 23859952.
- ↑ C.A. Heinlein, C. Chang. Androgen receptor in prostate cancer. „Endocr Rev”. 25 (2), s. 276–308, 2004. DOI: 10.1210/er.2002-0032. PMID: 15082523.
- ↑ P. Koivisto, J. Kononen, C. Palmberg, T. Tammela i inni. Androgen receptor gene amplification: a possible molecular mechanism for androgen deprivation therapy failure in prostate cancer. „Cancer Res”. 57 (2), s. 314–319, 1997. PMID: 9000575.
- ↑ H. Zhu, J.A. Garcia. Targeting the adrenal gland in castration-resistant prostate cancer: a case for orteronel, a selective CYP-17 17,20-lyase inhibitor. „Curr Oncol Rep”. 15 (2), s. 105–112, 2013. DOI: 10.1007/s11912-013-0300-1. PMID: 23371447.
- ↑ N. Mitsiades, C.C. Sung, N. Schultz, D.C. Danila i inni. Distinct patterns of dysregulated expression of enzymes involved in androgen synthesis and metabolism in metastatic prostate cancer tumors. „Cancer Res”. 72 (23), s. 6142–6152, 2012. DOI: 10.1158/0008-5472.CAN-12-1335. PMID: 22971343.
- ↑ a b S. Phin, M.W. Moore, P.D. Cotter. Genomic Rearrangements of PTEN in Prostate Cancer. „Front Oncol”. 3, s. 240, 2013. DOI: 10.3389/fonc.2013.00240. PMID: 24062990.
- ↑ a b c M.A. De Velasco, H. Uemura. Preclinical Remodeling of Human Prostate Cancer through the PTEN/AKT Pathway. „Adv Urol”. 2012, 2012. DOI: 10.1155/2012/419348. PMID: 22454635.
- ↑ a b c d e G. Khemlina, S. Ikeda, R. Kurzrock. Molecular landscape of prostate cancer: implications for current clinical trials. „Cancer Treat Rev”. 41 (9), s. 761–766, 2015. DOI: 10.1016/j.ctrv.2015.07.001. PMID: 26210103.
- ↑ a b C. Thangavel, E. Boopathi, Y. Liu, A. Haber i inni. RB Loss Promotes Prostate Cancer Metastasis. „Cancer Res”. 77 (4), s. 982–995, 2017. DOI: 10.1158/0008-5472.CAN-16-1589. PMID: 27923835.
- ↑ a b K.F. Macleod. The RB tumor suppressor: a gatekeeper to hormone independence in prostate cancer?. „J Clin Invest”. 120 (12), s. 4179–4182, 2010. DOI: 10.1172/JCI45406. PMID: 21099103.
- ↑ D. Hawksworth, L. Ravindranath, Y. Chen, B. Furusato i inni. Overexpression of C-MYC oncogene in prostate cancer predicts biochemical recurrence. „Prostate Cancer Prostatic Dis”. 13 (4), s. 311–315, 2010. DOI: 10.1038/pcan.2010.31. PMID: 20820186.
- ↑ C.M. Koh, C.J. Bieberich, C.V. Dang, W.G. Nelson i inni. MYC and Prostate Cancer. „Genes Cancer”. 1 (6), s. 617–628, 2010. DOI: 10.1177/1947601910379132. PMID: 21779461.
- ↑ a b Y. Wang, Y.X. Zhang, C.Z. Kong, Z. Zhang i inni. Loss of P53 facilitates invasion and metastasis of prostate cancer cells. „Mol Cell Biochem”. 384 (1–2), s. 121–127, 2013. DOI: 10.1007/s11010-013-1789-1. PMID: 23982184.
- ↑ a b E. Mazaris, A. Tsiotras. Molecular pathways in prostate cancer. „Nephrourol Mon”. 5 (3), s. 792–800, 2013. DOI: 10.5812/numonthly.9430. PMID: 24282788.
- ↑ DeVita, Lawrence i Rosenberg 2015 ↓, s. 930.
- ↑ a b K.H. Kim, C.W. Roberts. Targeting EZH2 in cancer. „Nat Med”. 22 (2), s. 128–134, 2016. DOI: 10.1038/nm.4036. PMID: 26845405.
- ↑ A.P. Bracken, D. Pasini, M. Capra, E. Prosperini i inni. EZH2 is downstream of the pRB-E2F pathway, essential for proliferation and amplified in cancer. „EMBO J”. 22 (20), s. 5323–5335, 2003. DOI: 10.1093/emboj/cdg542. PMID: 14532106.
- ↑ S.W. Bruggeman, M.E. Valk-Lingbeek, P.P. van der Stoop, J.J. Jacobs i inni. Ink4a and Arf differentially affect cell proliferation and neural stem cell self-renewal in Bmi1-deficient mice. „Genes Dev”. 19 (12), s. 1438–1443, 2005. DOI: 10.1101/gad.1299305. PMID: 15964995.
- ↑ Q. Cao, J. Yu, S.M. Dhanasekaran, J.H. Kim i inni. Repression of E-cadherin by the polycomb group protein EZH2 in cancer. „Oncogene”. 27 (58), s. 7274–7284, 2008. DOI: 10.1038/onc.2008.333. PMID: 18806826.
- ↑ J. Du, L. Li, Z. Ou, C. Kong i inni. FOXC1, a target of polycomb, inhibits metastasis of breast cancer cells. „Breast Cancer Res Treat”. 131 (1), s. 65–73, 2012. DOI: 10.1007/s10549-011-1396-3. PMID: 21465172.
- ↑ Y. Zhao, D.J. Tindall, H. Huang. Modulation of androgen receptor by FOXA1 and FOXO1 factors in prostate cancer. „Int J Biol Sci”. 10 (6), s. 614–619, 2014. DOI: 10.7150/ijbs.8389. PMID: 24948874.
- ↑ B. Sahu, M. Laakso, K. Ovaska, T. Mirtti i inni. Dual role of FoxA1 in androgen receptor binding to chromatin, androgen signalling and prostate cancer. „EMBO J”. 30 (19), s. 3962–3976, 2011. DOI: 10.1038/emboj.2011.328. PMID: 21915096.
- ↑ J. Gerhardt, M. Montani, P. Wild, M. Beer i inni. FOXA1 promotes tumor progression in prostate cancer and represents a novel hallmark of castration-resistant prostate cancer. „Am J Pathol”. 180 (2), s. 848–861, 2012. DOI: 10.1016/j.ajpath.2011.10.021. PMID: 22138582.
- ↑ R.K. Jain, R.J. Mehta, H. Nakshatri, M.T. Idrees i inni. High-level expression of forkhead-box protein A1 in metastatic prostate cancer. „Histopathology”. 58 (5), s. 766–772, 2011. DOI: 10.1111/j.1365-2559.2011.03796.x. PMID: 21401706.
- ↑ a b Y.A. Yang, J. Yu. Current perspectives on FOXA1 regulation of androgen receptor signaling and prostate cancer. „Genes Dis”. 2 (2), s. 144–151, 2015. DOI: 10.1016/j.gendis.2015.01.003. PMID: 26114156.
- ↑ C.E. Barbieri, S.A. Tomlins. The prostate cancer genome: perspectives and potential. „Urol Oncol”. 32 (1), 2014. DOI: 10.1016/j.urolonc.2013.08.025. PMID: 24239470.
- ↑ a b M.A. Augello, T.E. Hickey, K.E. Knudsen. FOXA1: master of steroid receptor function in cancer. „EMBO J”. 30 (19), s. 3885–3894, 2011. DOI: 10.1038/emboj.2011.340. PMID: 21934649.
- ↑ M. Blattner, D.J. Lee, C. O’Reilly, K. Park i inni. SPOP mutations in prostate cancer across demographically diverse patient cohorts. „Neoplasia”. 16 (1), s. 14–20, 2014. PMID: 24563616.
- ↑ a b C.E. Barbieri, S.C. Baca, M.S. Lawrence, F. Demichelis i inni. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. „Nat Genet”. 44 (6), s. 685–689, 2012. DOI: 10.1038/ng.2279. PMID: 22610119.
- ↑ J.P. Theurillat, N.D. Udeshi, W.J. Errington, T. Svinkina i inni. Prostate cancer. Ubiquitylome analysis identifies dysregulation of effector substrates in SPOP-mutant prostate cancer. „Science”. 346 (6205), s. 85–89, 2014. DOI: 10.1126/science.1250255. PMID: 25278611.
- ↑ W. Gan, X. Dai, A. Lunardi, Z. Li i inni. SPOP Promotes Ubiquitination and Degradation of the ERG Oncoprotein to Suppress Prostate Cancer Progression. „Mol Cell”. 59 (6), s. 917–930, 2015. DOI: 10.1016/j.molcel.2015.07.026. PMID: 26344095.
- ↑ M. Blattner, D. Liu, B.D. Robinson, D. Huang i inni. SPOP Mutation Drives Prostate Tumorigenesis In Vivo through Coordinate Regulation of PI3K/mTOR and AR Signaling. „Cancer Cell”. 31 (3), s. 436–451, 2017. DOI: 10.1016/j.ccell.2017.02.004. PMID: 28292441.
- ↑ G. Boysen, C.E. Barbieri, D. Prandi, M. Blattner i inni. SPOP mutation leads to genomic instability in prostate cancer. „Elife”. 4, 2015. DOI: 10.7554/eLife.09207. PMID: 26374986.
- ↑ a b c V. Kari, W.Y. Mansour, S.K. Raul, S.J. Baumgart i inni. Loss of CHD1 causes DNA repair defects and enhances prostate cancer therapeutic responsiveness. „EMBO Rep”. 17 (11), s. 1609–1623, 2016. DOI: 10.15252/embr.201642352. PMID: 27596623.
- ↑ Krzakowski i in. 2015 ↓, s. 740–742.
- ↑ J.P. Richie, W.J. Catalona, F.R. Ahmann, M.A. Hudson i inni. Effect of patient age on early detection of prostate cancer with serum prostate-specific antigen and digital rectal examination. „Urology”. 42 (4), s. 365–374, 1993. PMID: 7692657.
- ↑ G.F. Carvalhal, D.S. Smith, D.E. Mager, C. Ramos i inni. Digital rectal examination for detecting prostate cancer at prostate specific antigen levels of 4 ng./ml. or less. „J Urol”. 161 (3), s. 835–859, 1999. PMID: 10022696.
- ↑ a b c d e f g h i j k l m n o p N. Borley, M.R. Feneley. Prostate cancer: diagnosis and staging. „Asian J Androl”. 11 (1), s. 74–80, 2009. DOI: 10.1038/aja.2008.19. PMID: 19050692.
- ↑ a b c d H. Lilja, D. Ulmert, A.J. Vickers. Prostate-specific antigen and prostate cancer: prediction, detection and monitoring. „Nat Rev Cancer”. 8 (4), s. 268–278, 2008. DOI: 10.1038/nrc2351. PMID: 18337732.
- ↑ a b P.A. Humphrey, G.L. Andriole. Prostate cancer diagnosis. „Mo Med”. 107 (2). s. 107–112. PMID: 20446517.
- ↑ a b Mottet i in. 2017 ↓, s. 18–19.
- ↑ Thompson, I.M., et al. Prevalence of prostate cancer among men with a prostate-specific antigen level < or =4.0 ng per milliliter. N Engl J Med, 2004. 350: 2239.
- ↑ J.E. Oesterling, S.J. Jacobsen, C.G. Chute, H.A. Guess i inni. Serum prostate-specific antigen in a community-based population of healthy men. Establishment of age-specific reference ranges. „JAMA”. 270 (7), s. 860–864, 1993. PMID: 7688054.
- ↑ a b DeVita, Lawrence i Rosenberg 2015 ↓, s. 939–940.
- ↑ M. Ohori, J.K. Dunn, P.T. Scardino. Is prostate-specific antigen density more useful than prostate-specific antigen levels in the diagnosis of prostate cancer?. „Urology”. 46 (5), s. 666–671, 1995. DOI: 10.1016/S0090-4295(99)80298-2. PMID: 7495118.
- ↑ a b c d e f Mottet i in. 2017 ↓, s. 19.
- ↑ a b W.J. Catalona, A.W. Partin, K.M. Slawin, M.K. Brawer i inni. Use of the percentage of free prostate-specific antigen to enhance differentiation of prostate cancer from benign prostatic disease: a prospective multicenter clinical trial. „JAMA”. 279 (19), s. 1542–1547, 1998. PMID: 9605898.
- ↑ a b c d e f g h DeVita, Lawrence i Rosenberg 2015 ↓, s. 940.
- ↑ I.L. Deras, S.M. Aubin, A. Blase, J.R. Day i inni. PCA3: a molecular urine assay for predicting prostate biopsy outcome. „J Urol”. 179 (4), s. 1587–1592, 2008. DOI: 10.1016/j.juro.2007.11.038. PMID: 18295257.
- ↑ a b Mottet i in. 2017 ↓, s. 20.
- ↑ a b c Agnieszka Chomicz, Artur A. Antoniewicz. Fuzja krok po kroku. Przezkroczowa biopsja fuzyjna stercza z softwarową konsolidacją obrazów mpMRI i TRUS w czasie rzeczywistym przy użyciu BioJet System. „Przegląd Urologiczny”, 2017. [zarchiwizowane z adresu].
- ↑ a b c d e f g h Andrzej Lewicki, Stanisław Szempliński, Magdalena Zagrodzka, Tomasz Lorenc. Jak poprawić wyniki biopsji stercza. Zastosowanie wieloparametrycznego rezonansu magnetycznego oraz fuzji obrazów MRI i TRUS. „Przegląd Urologiczny”, 2017. [zarchiwizowane z adresu].
- ↑ a b c S.F. Shariat, C.G. Roehrborn. Using biopsy to detect prostate cancer. „Rev Urol”. 10 (4), s. 262–280, 2008. PMID: 19145270.
- ↑ a b c M.A. Bjurlin, S.S. Taneja. Standards for prostate biopsy. „Curr Opin Urol”. 24 (2), s. 155–161, 2014. DOI: 10.1097/MOU.0000000000000031. PMID: 24451092.
- ↑ A. de la Taille, P. Antiphon, L. Salomon, M. Cherfan i inni. Prospective evaluation of a 21-sample needle biopsy procedure designed to improve the prostate cancer detection rate. „Urology”. 61 (6), s. 1181–1186, 2003. PMID: 12809894.
- ↑ K. Eichler, S. Hempel, J. Wilby, L. Myers i inni. Diagnostic value of systematic biopsy methods in the investigation of prostate cancer: a systematic review. „J Urol”. 175 (5), s. 1605–1612, 2006. DOI: 10.1016/S0022-5347(05)00957-2. PMID: 16600713.
- ↑ S.S. Dianat, H.B. Carter, K.J. Macura. Magnetic Resonance-Guided Prostate Biopsy. „Magn Reson Imaging Clin N Am”. 23 (4), s. 621–631, 2015. DOI: 10.1016/j.mric.2015.05.005. PMID: 26499279.
- ↑ C.K. Kim. Magnetic resonance imaging-guided prostate biopsy: present and future. „Korean J Radiol”. 16 (1). s. 90–98. DOI: 10.3348/kjr.2015.16.1.90. PMID: 25598677.
- ↑ a b c d e f g h i Mottet i in. 2017 ↓, s. 22.
- ↑ A. van Hove, P.H. Savoie, C. Maurin, S. Brunelle i inni. Comparison of image-guided targeted biopsies versus systematic randomized biopsies in the detection of prostate cancer: a systematic literature review of well-designed studies. „World J Urol”. 32 (4), s. 847–858, 2014. DOI: 10.1007/s00345-014-1332-3. PMID: 24919965.
- ↑ I.G. Schoots, M.J. Roobol, D. Nieboer, C.H. Bangma i inni. Magnetic resonance imaging-targeted biopsy may enhance the diagnostic accuracy of significant prostate cancer detection compared to standard transrectal ultrasound-guided biopsy: a systematic review and meta-analysis. „Eur Urol”. 68 (3), s. 438–450, 2015. DOI: 10.1016/j.eururo.2014.11.037. PMID: 25480312.
- ↑ M. Valerio, I. Donaldson, M. Emberton, B. Ehdaie i inni. Detection of Clinically Significant Prostate Cancer Using Magnetic Resonance Imaging-Ultrasound Fusion Targeted Biopsy: A Systematic Review. „Eur Urol”. 68 (1), s. 8–19, 2015. DOI: 10.1016/j.eururo.2014.10.026. PMID: 25454618.
- ↑ M.M. Siddiqui, S. Rais-Bahrami, B. Turkbey, A.K. George i inni. Comparison of MR/ultrasound fusion-guided biopsy with ultrasound-guided biopsy for the diagnosis of prostate cancer. „JAMA”. 313 (4), s. 390–397, 2015. DOI: 10.1001/jama.2014.17942. PMID: 25626035.
- ↑ O. Wegelin, H.H. E. van Melick, L. Hooft, J. Bosch i inni. Comparing Three Different Techniques for Magnetic Resonance Imaging-targeted Prostate Biopsies: A Systematic Review of In-bore versus Magnetic Resonance Imaging-transrectal Ultrasound fusion versus Cognitive Registration. Is There a Preferred Technique?. „Eur Urol”. 71 (4), s. 517–531, 2017. DOI: 10.1016/j.eururo.2016.07.041. PMID: 27568655.
- ↑ J. Wu, A. Ji, B. Xie, X. Wang i inni. Is magnetic resonance/ultrasound fusion prostate biopsy better than systematic prostate biopsy? An updated meta- and trial sequential analysis. „Oncotarget”. 6 (41), 2015. DOI: 10.18632/oncotarget.6201. PMID: 26498362.
- ↑ V. Panebianco, F. Barchetti, A. Sciarra, A. Ciardi i inni. Multiparametric magnetic resonance imaging vs. standard care in men being evaluated for prostate cancer: a randomized study. „Urol Oncol”. 33 (1), 2015. DOI: 10.1016/j.urolonc.2014.09.013. PMID: 25443268.
- ↑ E. Baco, E. Rud, L.M. Eri, G. Moen i inni. A Randomized Controlled Trial To Assess and Compare the Outcomes of Two-core Prostate Biopsy Guided by Fused Magnetic Resonance and Transrectal Ultrasound Images and Traditional 12-core Systematic Biopsy. „Eur Urol”. 69 (1), s. 149–156, 2016. DOI: 10.1016/j.eururo.2015.03.041. PMID: 25862143.
- ↑ P.P. Tonttila, J. Lantto, E. Pääkkö, U. Piippo i inni. Prebiopsy Multiparametric Magnetic Resonance Imaging for Prostate Cancer Diagnosis in Biopsy-naive Men with Suspected Prostate Cancer Based on Elevated Prostate-specific Antigen Values: Results from a Randomized Prospective Blinded Controlled Trial. „Eur Urol”. 69 (3), s. 419–425, 2016. DOI: 10.1016/j.eururo.2015.05.024. PMID: 26033153.
- ↑ a b c Mottet i in. 2017 ↓, s. 23.
- ↑ K.A. Roehl, J.A. Antenor, W.J. Catalona. Serial biopsy results in prostate cancer screening study. „J Urol”. 167 (6), s. 2435–2439, 2002. PMID: 11992052.
- ↑ B. Djavan, V. Ravery, A. Zlotta, P. Dobronski i inni. Prospective evaluation of prostate cancer detected on biopsies 1, 2, 3 and 4: when should we stop?. „J Urol”. 166 (5), s. 1679–1683, 2001. PMID: 11586201.
- ↑ A., Nicholson i inni, The clinical effectiveness and cost-effectiveness of the PROGENSA(R) prostate cancer antigen 3 assay and the Prostate Health Index in the diagnosis of prostate cancer: a systematic review and economic evaluation. Health Technol Assess, 2015. 19: 1.
- ↑ a b Mottet i in. 2017 ↓, s. 21.
- ↑ Adam Gołąb. Prospektywna porównawcza ocena pierwszorazowej biopsji stercza wykonanej zmodyfikowaną techniką sekstantową i saturacyjną. „Przegląd Urologiczny”, 2008.
- ↑ B.R. Lane, C.D. Zippe, R. Abouassaly, L. Schoenfield i inni. Saturation technique does not decrease cancer detection during followup after initial prostate biopsy. „J Urol”. 179 (5), s. 1746–1750, 2008. DOI: 10.1016/j.juro.2008.01.049. PMID: 18343412.
- ↑ Mohler i in. 2017 ↓, s. 22.
- ↑ Mohler i in. 2017 ↓, s. 45.
- ↑ a b c d e f g h i j E.K. Outwater, J.L. Montilla-Soler. Imaging of prostate carcinoma. „Cancer Control”. 20 (3), s. 161–176, Jul 2013. DOI: 10.1177/107327481302000304. PMID: 23811700.
- ↑ a b c C.J. Harvey, J. Pilcher, J. Richenberg, U. Patel i inni. Applications of transrectal ultrasound in prostate cancer. „Br J Radiol”. 85 Spec No 1, 2012. DOI: 10.1259/bjr/56357549. PMID: 22844031.
- ↑ a b DeVita, Lawrence i Rosenberg 2015 ↓, s. 941.
- ↑ a b c d e f S. Sharma. Imaging and intervention in prostate cancer: Current perspectives and future trends. „Indian J Radiol Imaging”. 24 (2), s. 139–148, 2014. DOI: 10.4103/0971-3026.134399. PMID: 25024523.
- ↑ a b c d e f g h i S. Sarkar, S. Das. A Review of Imaging Methods for Prostate Cancer Detection. „Biomed Eng Comput Biol”. 7 (Suppl 1), s. 1–15, 2016. DOI: 10.4137/BECB.S34255. PMID: 26966397.
- ↑ F. Bratan, E. Niaf, C. Melodelima, A.L. Chesnais i inni. Influence of imaging and histological factors on prostate cancer detection and localisation on multiparametric MRI: a prospective study. „Eur Radiol”. 23 (7), s. 2019–2029, 2013. DOI: 10.1007/s00330-013-2795-0. PMID: 23494494.
- ↑ a b c d e f g Katarzyna Sklinda, Agnieszka Dąbrowska, Paweł Olejnik, Jerzy Walecki. Badanie multiparametryczne MRI (mpMRI) w PI-RADS v2. Komentarz dotyczący sposobu przeprowadzania badania multiparametrycznego MRI w PI-RADS v2. Część 3.. „Przegląd Urologiczny”, 2017. [zarchiwizowane z adresu].
- ↑ a b S. Verma, B. Turkbey, N. Muradyan, A. Rajesh i inni. Overview of dynamic contrast-enhanced MRI in prostate cancer diagnosis and management. „AJR Am J Roentgenol”. 198 (6), s. 1277–1288, 2012. DOI: 10.2214/AJR.12.8510. PMID: 22623539.
- ↑ a b B. Li, Y. Du, H. Yang, Y. Huang i inni. Magnetic resonance imaging for prostate cancer clinical application. „Chin J Cancer Res”. 25 (2), s. 240–249, 2013. DOI: 10.3978/j.issn.1000-9604.2013.03.06. PMID: 23592906.
- ↑ a b c Mottet i in. 2017 ↓, s. 27.
- ↑ M. de Rooij, E.H. Hamoen, J.A. Witjes, J.O. Barentsz i inni. Accuracy of Magnetic Resonance Imaging for Local Staging of Prostate Cancer: A Diagnostic Meta-analysis. „Eur Urol”. 70 (2), s. 233–245, 2016. DOI: 10.1016/j.eururo.2015.07.029. PMID: 26215604.
- ↑ a b c Mottet i in. 2017 ↓, s. 28.
- ↑ G. Shen, H. Deng, S. Hu, Z. Jia. Comparison of choline-PET/CT, MRI, SPECT, and bone scintigraphy in the diagnosis of bone metastases in patients with prostate cancer: a meta-analysis. „Skeletal Radiol”. 43 (11), s. 1503–1513, 2014. DOI: 10.1007/s00256-014-1903-9. PMID: 24841276.
- ↑ E. Even-Sapir, U. Metser, E. Mishani, G. Lievshitz i inni. The detection of bone metastases in patients with high-risk prostate cancer: 99mTc-MDP Planar bone scintigraphy, single- and multi-field-of-view SPECT, 18F-fluoride PET, and 18F-fluoride PET/CT. „J Nucl Med”. 47 (2), s. 287–297, 2006. PMID: 16455635.
- ↑ a b c d e f g h K.L. Wallitt, S.R. Khan, S. Dubash, H.H. Tam i inni. Clinical PET Imaging in Prostate Cancer. „Radiographics”. 37 (5). s. 1512–1536. DOI: 10.1148/rg.2017170035. PMID: 28800286.
- ↑ L. Evangelista, M. Cimitan, F. Zattoni, A. Guttilla i inni. Comparison between conventional imaging (abdominal-pelvic computed tomography and bone scan) and [(18)F]choline positron emission tomography/computed tomography imaging for the initial staging of patients with intermediate- tohigh-risk prostate cancer: A retrospective analysis. „Scand J Urol”. 49 (5), s. 345–353, 2015. DOI: 10.3109/21681805.2015.1005665. PMID: 25649494.
- ↑ F.E. von Eyben, K. Kairemo. Meta-analysis of (11)C-choline and (18)F-choline PET/CT for management of patients with prostate cancer. „Nucl Med Commun”. 35 (3), s. 221–230, 2014. DOI: 10.1097/MNM.0000000000000040. PMID: 24240194.
- ↑ Fanti S, Minozzi S, Castellucci P, Balduzzi S, Herrmann K, Krause BJ, et al. PET/CT with (11)C-choline for evaluation of prostate cancer patients with biochemical recurrence: meta-analysis and critical review of available data. Eur J Nucl Med Mol Imaging. 2016;43:55–69.
- ↑ Treglia G, Annunziata S, Pizzuto DA, Giovanella L, Prior JO, Ceriani L. Detection Rate of 18F-Labeled PSMA PET/CT in Biochemical Recurrent Prostate Cancer: A Systematic Review and a Meta-Analysis. Cancers. 2019;11.
- ↑ Cornford P, Bellmunt J, Bolla M, Briers E, De Santis M, Gross T, et al. EAU-ESTRO-SIOG Guidelines on Prostate Cancer. Part II: Treatment of Relapsing, Metastatic, and Castration-Resistant Prostate Cancer. Eur Urol. 2017;71:630–42.
- ↑ U. Tateishi, S. Morita, M. Taguri, K. Shizukuishi i inni. A meta-analysis of (18)F-Fluoride positron emission tomography for assessment of metastatic bone tumor. „Ann Nucl Med”. 24 (7), s. 523–531, 2010. DOI: 10.1007/s12149-010-0393-7. PMID: 20559896.
- ↑ Udovicich C i inni, 68 Ga-prostate-specific membrane antigen-positron emission tomography/computed tomography in advanced prostate cancer: Current state and future trends, „Prostate Int”, 2017, DOI: 10.1016/j.prnil.2017.02.003.
- ↑ Afshar-Oromieh A, Malcher A, Eder M, et al. PET imaging with a [68Ga] gallium-labelled PSMA ligand for the diagnosis of prostate cancer: biodistribution in humans and first evaluation of tumour lesions. Eur J Nucl Med Mol Imaging, 2013; 40 (4): 486–495.
- ↑ Cardinale J, Schäfer M, Benešová M, et al. Preclinical Evaluation of 18F-PSMA-1007, a New Prostate-Specific Membrane Antigen Ligand for Prostate Cancer Imaging. J Nucl Med, 2017; 58 (3): 425–431.
- ↑ Sanchez-Crespo A. Comparison of Gallium-68 and Fluorine-18 imaging characteristics in positron emission tomography. Appl Radiat Isot, 2013; 76: 55–62.
- ↑ a b Giesel FL, Hadaschik B, Cardinale J, et al. F-18 labelled PSMA-1007: biodistribution, radiation dosimetry and histopathological validation of tumor lesions in prostate cancer patients. Eur J Nucl Med Mol Imaging, 2017; 44 (4): 678–688.
- ↑ Benešová M, Bauder-Wüst U, Schäfer M, et al. Linker Modification Strategies To Control the Prostate-Specific Membrane Antigen (PSMA)-Targeting and Pharmacokinetic Properties of DOTA-Conjugated PSMA Inhibitors. J Med Chem, 2016; 59 (5): 1761–1775.
- ↑ Giesel FL, Will L, Paddubny K, et al. [18F]PSMA-1007 PET Improves the Diagnosis of Local Recurrence and Lymph Node Metastases in a Prostate Cancer Patient With a History of Bilateral Hip Arthroplasty. Clin Genitourin Cancer, 2018; 16 (2): 111–113.
- ↑ Giesel FL, Knorr K, Spohn F, et al. Detection efficacy of [18 F]PSMA-1007 PET/CT in 251 Patients with biochemical recurrence after radical prostatectomy. J Nucl Med, 2018: jnumed.118.212233.
- ↑ Rahbar K, Afshar-Oromieh A, Seifert R, et al. Diagnostic performance of 18F-PSMA-1007 PET/CT in patients with biochemical recurrent prostate cancer. Eur J Nucl Med Mol Imaging, 2018; 45 (12): 2055–2061.
- ↑ Morigi JJ, Stricker PD, van Leeuwen PJ, et al. Prospective Comparison of 18F-Fluoromethylcholine Versus 68Ga-PSMA PET/CT in Prostate Cancer Patients Who Have Rising PSA After Curative Treatment and Are Being Considered for Targeted Therapy. J Nucl Med, 2015; 56 (8): 1185–1190.
- ↑ Giesel FL, Will L, Lawal I, et al. Intraindividual Comparison of 18F-PSMA-1007 and 18 F-DCFPyL PET/CT in the Prospective Evaluation of Patients with Newly Diagnosed Prostate Carcinoma: A Pilot Study. J Nucl Med, 2018; 59 (7): 1076–1080.
- ↑ Rutkowski i Warzocha 2013 ↓, s. 337–338.
- ↑ Mottet i in. 2017 ↓, s. 29.
- ↑ a b Mottet i in. 2017 ↓, s. 15.
- ↑ a b Mohler i in. 2017 ↓, s. 37.
- ↑ Krzakowski i in. 2015 ↓, s. 743.
- ↑ a b V. Pagliarulo, S. Bracarda, M.A. Eisenberger, N. Mottet i inni. Contemporary role of androgen deprivation therapy for prostate cancer. „Eur Urol”. 61 (1), s. 11–25, 2012. DOI: 10.1016/j.eururo.2011.08.026. PMID: 21871711.
- ↑ a b M.R. Robinson, P.H. Smith, B. Richards, D.W. Newling i inni. The final analysis of the EORTC Genito-Urinary Tract Cancer Co-Operative Group phase III clinical trial (protocol 30805) comparing orchidectomy, orchidectomy plus cyproterone acetate and low dose stilboestrol in the management of metastatic carcinoma of the prostate. „Eur Urol”. 28 (4), s. 273–283, 1995. PMID: 8575493.
- ↑ a b J. Seidenfeld, D.J. Samson, V. Hasselblad, N. Aronson i inni. Single-therapy androgen suppression in men with advanced prostate cancer: a systematic review and meta-analysis. „Ann Intern Med”. 132 (7), s. 566–577, 2000. PMID: 10744594.
- ↑ EAU Guidelines. Edn. presented at the EAU Annual Congress Amsterdam 2020. ISBN 978-94-92671-07-3. https://uroweb.org/guideline/prostate-cancer/.
- ↑ Opracowanie analityczne AOTMiT. Kompleksowa opieka onkologiczna – model organizacji diagnostyki i leczenia raka gruczołu krokowego. WS.4320.1.2019.
- ↑ a b c d DeVita, Lawrence i Rosenberg 2015 ↓, s. 944.
- ↑ a b c d e Mottet i in. 2017 ↓, s. 30.
- ↑ a b c d e Mohler i in. 2017 ↓, s. 48.
- ↑ a b Mohler i in. 2017 ↓, s. 73.
- ↑ a b Mohler i in. 2017 ↓, s. 74.
- ↑ Mohler i in. 2017 ↓, s. 75.
- ↑ a b DeVita, Lawrence i Rosenberg 2015 ↓, s. 961.
- ↑ a b Krzakowski i in. 2015 ↓, s. 746–747.
- ↑ Mottet i in. 2017 ↓, s. 82–83.
- ↑ a b Krzakowski i in. 2015 ↓, s. 749.
- ↑ a b Rutkowski i Warzocha 2013 ↓, s. 340.
- ↑ a b M.S. Chung, S.H. Lee. Current status of active surveillance in prostate cancer. „Investig Clin Urol”. 57 (1), s. 14–20, 2016. DOI: 10.4111/icu.2016.57.1.14. PMID: 26966722.
- ↑ a b Mottet i in. 2017 ↓, s. 34.
- ↑ J.H. Wang, T.M. Downs, E. Jason Abel, K.A. Richards i inni. Prostate Biopsy in Active Surveillance Protocols: Immediate Re-biopsy and Timing of Subsequent Biopsies. „Curr Urol Rep”. 18 (7), s. 48, 2017. DOI: 10.1007/s11934-017-0702-y. PMID: 28589399.
- ↑ J.D. Garisto, L. Klotz. Active Surveillance for Prostate Cancer: How to Do It Right. „Oncology (Williston Park)”. 31 (5), s. 333–340, 2017. PMID: 28512731.
- ↑ a b Mottet i in. 2017 ↓, s. 31.
- ↑ F.B. Thomsen, K. Brasso, L.H. Klotz, M.A. Røder i inni. Active surveillance for clinically localized prostate cancer-a systematic review. „J Surg Oncol”. 109 (8), s. 830–835, 2014. DOI: 10.1002/jso.23584. PMID: 24610744.
- ↑ L. Klotz, D. Vesprini, P. Sethukavalan, V. Jethava i inni. Long-term follow-up of a large active surveillance cohort of patients with prostate cancer. „J Clin Oncol”. 33 (3), s. 272–277, Jan 2015. DOI: 10.1200/JCO.2014.55.1192. PMID: 25512465.
- ↑ a b F.C. Hamdy, J.L. Donovan, J.A. Lane, M. Mason i inni. 10-Year Outcomes after Monitoring, Surgery, or Radiotherapy for Localized Prostate Cancer. „N Engl J Med”. 375 (15), s. 1415–1424, 10 2016. DOI: 10.1056/NEJMoa1606220. PMID: 27626136.
- ↑ S. Loeb, Y. Folkvaljon, D.V. Makarov, O. Bratt i inni. Five-year nationwide follow-up study of active surveillance for prostate cancer. „Eur Urol”. 67 (2), s. 233–238, 2015. DOI: 10.1016/j.eururo.2014.06.010. PMID: 24993868.
- ↑ S. Roemeling, M.J. Roobol, S.H. de Vries, T. Wolters i inni. Active surveillance for prostate cancers detected in three subsequent rounds of a screening trial: characteristics, PSA doubling times, and outcome. „Eur Urol”. 51 (5), s. 1244–1250, 2007. DOI: 10.1016/j.eururo.2006.11.053. PMID: 17161520.
- ↑ J.J. Tosoian, M. Mamawala, J.I. Epstein, P. Landis i inni. Intermediate and Longer-Term Outcomes From a Prospective Active-Surveillance Program for Favorable-Risk Prostate Cancer. „J Clin Oncol”. 33 (30), s. 3379–3385, 2015. DOI: 10.1200/JCO.2015.62.5764. PMID: 26324359.
- ↑ N.J. van As, A.R. Norman, K. Thomas, V.S. Khoo i inni. Predicting the probability of deferred radical treatment for localised prostate cancer managed by active surveillance. „Eur Urol”. 54 (6), s. 1297–1305, 2008. DOI: 10.1016/j.eururo.2008.02.039. PMID: 18342430.
- ↑ A.J. Simpkin, K. Tilling, R.M. Martin, J.A. Lane i inni. Systematic Review and Meta-analysis of Factors Determining Change to Radical Treatment in Active Surveillance for Localized Prostate Cancer. „Eur Urol”. 67 (6), s. 993–1005, 2015. DOI: 10.1016/j.eururo.2015.01.004. PMID: 25616709.
- ↑ S. Loeb, Q. Zhou, U. Siebert, U. Rochau i inni. Active Surveillance Versus Watchful Waiting for Localized Prostate Cancer: A Model to Inform Decisions. „Eur Urol”. 72 (6), s. 899–907, 2017. DOI: 10.1016/j.eururo.2017.07.018. PMID: 28844371.
- ↑ a b c Albala i in. 2011 ↓, s. 218.
- ↑ a b c d e DeVita, Lawrence i Rosenberg 2015 ↓, s. 945.
- ↑ Albala i in. 2011 ↓, s. 218–219.
- ↑ Blandy i Kaisary 2009 ↓, s. 196–197.
- ↑ Blandy i Kaisary 2009 ↓, s. 197.
- ↑ DeVita, Lawrence i Rosenberg 2015 ↓, s. 945–946.
- ↑ a b Mohler i in. 2017 ↓, s. 54.
- ↑ a b c d e f g Mottet i in. 2017 ↓, s. 36.
- ↑ M.E. Allaf, G.S. Palapattu, B.J. Trock, H.B. Carter i inni. Anatomical extent of lymph node dissection: impact on men with clinically localized prostate cancer. „J Urol”. 172, s. 1840–1844, 2004. PMID: 15540734.
- ↑ A. Heidenreich, Z. Varga, R. Von Knobloch. Extended pelvic lymphadenectomy in patients undergoing radical prostatectomy: high incidence of lymph node metastasis. „J Urol”. 167 (4), s. 1681–1686, 2002. PMID: 11912387.
- ↑ K.A. Touijer, C.R. Mazzola, D.D. Sjoberg, P.T. Scardino i inni. Long-term outcomes of patients with lymph node metastasis treated with radical prostatectomy without adjuvant androgen-deprivation therapy. „Eur Urol”. 65 (1), s. 20–25, 2014. DOI: 10.1016/j.eururo.2013.03.053. PMID: 23619390.
- ↑ C. von Bodman, G. Godoy, D.C. Chade, A. Cronin i inni. Predicting biochemical recurrence-free survival for patients with positive pelvic lymph nodes at radical prostatectomy. „J Urol”. 184 (1), s. 143–148, 2010. DOI: 10.1016/j.juro.2010.03.039. PMID: 20478587.
- ↑ N. Fossati, P.M. Willemse, T. Van den Broeck, R.C. N. van den Bergh i inni. The Benefits and Harms of Different Extents of Lymph Node Dissection During Radical Prostatectomy for Prostate Cancer: A Systematic Review. „Eur Urol”. 72 (1), s. 84–109, 2017. DOI: 10.1016/j.eururo.2016.12.003. PMID: 28126351.
- ↑ DeVita, Lawrence i Rosenberg 2015 ↓, s. 946.
- ↑ Mohler i in. 2017 ↓, s. 53–54.
- ↑ DeVita, Lawrence i Rosenberg 2015 ↓, s. 947.
- ↑ a b T.J. Wilt, M.K. Brawer, K.M. Jones, M.J. Barry i inni. Radical prostatectomy versus observation for localized prostate cancer. „N Engl J Med”. 367 (3), s. 203–213, 2012. DOI: 10.1056/NEJMoa1113162. PMID: 22808955.
- ↑ A. Bill-Axelson, L. Holmberg, H. Garmo, J.R. Rider i inni. Radical prostatectomy or watchful waiting in early prostate cancer. „N Engl J Med”. 370 (10), s. 932–942, 2014. DOI: 10.1056/NEJMoa1311593. PMID: 24597866.
- ↑ a b c Mottet i in. 2017 ↓, s. 35.
- ↑ Mohler i in. 2017 ↓, s. 52.
- ↑ S. Joniau, C.Y. Hsu, P. Gontero, M. Spahn i inni. Radical prostatectomy in very high-risk localized prostate cancer: long-term outcomes and outcome predictors. „Scand J Urol Nephrol”. 46 (3), s. 164–171, 2012. DOI: 10.3109/00365599.2011.637956. PMID: 22364377.
- ↑ P.A. Johnstone, K.C. Ward, M. Goodman, V. Assikis i inni. Radical prostatectomy for clinical T4 prostate cancer. „Cancer”. 106 (12), s. 2603–2609, 2006. DOI: 10.1002/cncr.21926. PMID: 16700037.
- ↑ F. Moltzahn, J. Karnes, P. Gontero, B. Kneitz i inni. Predicting prostate cancer-specific outcome after radical prostatectomy among men with very high-risk cT3b/4 PCa: a multi-institutional outcome study of 266 patients. „Prostate Cancer Prostatic Dis”. 18 (1), s. 31–37, 2015. DOI: 10.1038/pcan.2014.41. PMID: 25535100.
- ↑ Albala i in. 2011 ↓, s. 222–223.
- ↑ P. Iversen, D.G. McLeod, W.A. See, T. Morris i inni. Antiandrogen monotherapy in patients with localized or locally advanced prostate cancer: final results from the bicalutamide Early Prostate Cancer programme at a median follow-up of 9.7 years. „BJU Int”. 105 (8), s. 1074–1081, 2010. DOI: 10.1111/j.1464-410X.2010.09319.x. PMID: 22129214.
- ↑ S. Kumar, M. Shelley, C. Harrison, B. Coles i inni. Neo-adjuvant and adjuvant hormone therapy for localised and locally advanced prostate cancer. „Cochrane Database Syst Rev”, 2006. DOI: 10.1002/14651858.CD006019.pub2. PMID: 17054269.
- ↑ a b c d Krzakowski i in. 2015 ↓, s. 747.
- ↑ E.M. Messing, J. Manola, J. Yao, M. Kiernan i inni. Immediate versus deferred androgen deprivation treatment in patients with node-positive prostate cancer after radical prostatectomy and pelvic lymphadenectomy. „Lancet Oncol”. 7 (6), s. 472–479, 2006. DOI: 10.1016/S1470-2045(06)70700-8. PMID: 16750497.
- ↑ Mottet i in. 2017 ↓, s. 36–37.
- ↑ Mohler i in. 2017 ↓, s. 60.
- ↑ a b Mottet i in. 2017 ↓, s. 50.
- ↑ Mottet i in. 2017 ↓, s. 51.
- ↑ Marek Zawadzki, Stefan W. Czarniecki, Marek Filipek. Zastosowanie ogniskowanej wysokoenergetycznej terapii ultradźwiękowej (HIFU) w raku stercza. „Przegląd Urologiczny”, 2013.
- ↑ Mottet i in. 2017 ↓, s. 49–50.
- ↑ a b c d Mottet i in. 2017 ↓, s. 49.
- ↑ a b C.R. Ramsay, T.E. Adewuyi, J. Gray, J. Hislop i inni. Ablative therapy for people with localised prostate cancer: a systematic review and economic evaluation. „Health Technol Assess”. 19 (49), 2015. DOI: 10.3310/hta19490. PMID: 26140518.
- ↑ a b Mohler i in. 2017 ↓, s. 61.
- ↑ K.M. Siddiqui, M. Billia, A. Arifin, F. Li i inni. Pathological, Oncologic and Functional Outcomes of a Prospective Registry of Salvage High Intensity Focused Ultrasound Ablation for Radiorecurrent Prostate Cancer. „J Urol”. 197 (1), s. 97–102, 2017. DOI: 10.1016/j.juro.2016.06.092. PMID: 27422297.
- ↑ a b Krzakowski i in. 2015 ↓, s. 745.
- ↑ a b c DeVita, Lawrence i Rosenberg 2015 ↓, s. 951.
- ↑ a b c d DeVita, Lawrence i Rosenberg 2015 ↓, s. 956.
- ↑ a b c d e Mottet i in. 2017 ↓, s. 48.
- ↑ a b c d e Krzakowski i in. 2015 ↓, s. 746.
- ↑ Krzakowski i in. 2015 ↓, s. 98.
- ↑ a b Gunderson i Tepper 2012 ↓, s. 1059.
- ↑ a b Krzakowski i in. 2015 ↓, s. 100.
- ↑ DeVita, Lawrence i Rosenberg 2015 ↓, s. 951–952.
- ↑ V. Beckendorf, S. Guerif, E. Le Prisé, J.M. Cosset i inni. 70 Gy versus 80 Gy in localized prostate cancer: 5-year results of GETUG 06 randomized trial. „Int J Radiat Oncol Biol Phys”. 80 (4), s. 1056–1063, 2011. DOI: 10.1016/j.ijrobp.2010.03.049. PMID: 21147514.
- ↑ D.A. Kuban, S.L. Tucker, L. Dong, G. Starkschall i inni. Long-term results of the M. D. Anderson randomized dose-escalation trial for prostate cancer. „Int J Radiat Oncol Biol Phys”. 70 (1), s. 67–74, 2008. DOI: 10.1016/j.ijrobp.2007.06.054. PMID: 17765406.
- ↑ A.L. Zietman, K. Bae, J.D. Slater, W.U. Shipley i inni. Randomized trial comparing conventional-dose with high-dose conformal radiation therapy in early-stage adenocarcinoma of the prostate: long-term results from proton radiation oncology group/american college of radiology 95-09. „J Clin Oncol”. 28 (7), s. 1106–1111, 2010. DOI: 10.1200/JCO.2009.25.8475. PMID: 20124169.
- ↑ D.P. Dearnaley, G. Jovic, I. Syndikus, V. Khoo i inni. Escalated-dose versus control-dose conformal radiotherapy for prostate cancer: long-term results from the MRC RT01 randomised controlled trial. „Lancet Oncol”. 15 (4), s. 464–473, 2014. DOI: 10.1016/S1470-2045(14)70040-3. PMID: 24581940.
- ↑ D.A. Kuban, L.B. Levy, M.R. Cheung, A.K. Lee i inni. Long-term failure patterns and survival in a randomized dose-escalation trial for prostate cancer. Who dies of disease?. „Int J Radiat Oncol Biol Phys”. 79 (5), s. 1310–1317, 2011. DOI: 10.1016/j.ijrobp.2010.01.006. PMID: 20493642.
- ↑ A. Kalbasi, J. Li, A. Berman, S. Swisher-McClure i inni. Dose-Escalated Irradiation and Overall Survival in Men With Nonmetastatic Prostate Cancer. „JAMA Oncol”. 1 (7), s. 897–906, 2015. DOI: 10.1001/jamaoncol.2015.2316. PMID: 26181727.
- ↑ a b Mottet i in. 2017 ↓, s. 39.
- ↑ a b c Gunderson i Tepper 2012 ↓, s. 1061.
- ↑ a b c DeVita, Lawrence i Rosenberg 2015 ↓, s. 953.
- ↑ a b Mottet i in. 2017 ↓, s. 40.
- ↑ Y.T. Wachtfogel, W. Abrams, U. Kucich, G. Weinbaum i inni. Fibronectin degradation products containing the cytoadhesive tetrapeptide stimulate human neutrophil degranulation. „J Clin Invest”. 81 (5), s. 1310–1316, 1988. DOI: 10.1172/JCI113456. PMID: 2966812.
- ↑ L. Incrocci, R.C. Wortel, W.G. Alemayehu, S. Aluwini i inni. Hypofractionated versus conventionally fractionated radiotherapy for patients with localised prostate cancer (HYPRO): final efficacy results from a randomised, multicentre, open-label, phase 3 trial. „Lancet Oncol”. 17 (8), s. 1061–1069, 2016. DOI: 10.1016/S1470-2045(16)30070-5. PMID: 27339116.
- ↑ D. Dearnaley, I. Syndikus, H. Mossop, V. Khoo i inni. Conventional versus hypofractionated high-dose intensity-modulated radiotherapy for prostate cancer: 5-year outcomes of the randomised, non-inferiority, phase 3 CHHiP trial. „Lancet Oncol”. 17 (8), s. 1047–1060, 2016. DOI: 10.1016/S1470-2045(16)30102-4. PMID: 27339115.
- ↑ W.R. Lee, J.J. Dignam, M.B. Amin, D.W. Bruner i inni. Randomized Phase III Noninferiority Study Comparing Two Radiotherapy Fractionation Schedules in Patients With Low-Risk Prostate Cancer. „J Clin Oncol”. 34 (20), s. 2325–2332, 2016. DOI: 10.1200/JCO.2016.67.0448. PMID: 27044935.
- ↑ B.F. Koontz, A. Bossi, C. Cozzarini, T. Wiegel i inni. A systematic review of hypofractionation for primary management of prostate cancer. „Eur Urol”. 68 (4), s. 683–691, 2015. DOI: 10.1016/j.eururo.2014.08.009. PMID: 25171903.
- ↑ a b c d Mottet i in. 2017 ↓, s. 44.
- ↑ a b Mohler i in. 2017 ↓, s. 27.
- ↑ Gunderson i Tepper 2012 ↓, s. 141.
- ↑ a b Mohler i in. 2017 ↓, s. 56.
- ↑ a b DeVita, Lawrence i Rosenberg 2015 ↓, s. 954.
- ↑ a b c d Mottet i in. 2017 ↓, s. 46.
- ↑ a b c Allen M. Chen, Srinivasan Vijayakumar: Prostate Cancer. Demos Medical Publishing, 2011, s. 82–83. ISBN 978-1-61705-067-1.
- ↑ a b c Gunderson i Tepper 2012 ↓, s. 1091.
- ↑ a b c d Allen M. Chen, Srinivasan Vijayakumar: Prostate Cancer. Demos Medical Publishing, 2011, s. 88–89. ISBN 978-1-61705-067-1.
- ↑ F.M. Fang, Y.M. Wang, C.J. Wang, H.Y. Huang i inni. Comparison of the outcome and morbidity for localized or locally advanced prostate cancer treated by high-dose-rate brachytherapy plus external beam radiotherapy (EBRT) versus EBRT alone. „Jpn J Clin Oncol”. 38 (7), s. 474–479, 2008. DOI: 10.1093/jjco/hyn056. PMID: 18621848.
- ↑ O. Al-Salihi, A. Mitra, H. Payne. Challenge of dose escalation in locally advanced unfavourable prostate cancer using HDR brachytherapy. „Prostate Cancer Prostatic Dis”. 9 (4), s. 370–373, 2006. DOI: 10.1038/sj.pcan.4500893. PMID: 16832383.
- ↑ a b Mohler i in. 2017 ↓, s. 57.
- ↑ P.J. Hoskin, A.M. Rojas, P.J. Bownes, G.J. Lowe i inni. Randomised trial of external beam radiotherapy alone or combined with high-dose-rate brachytherapy boost for localised prostate cancer. „Radiother Oncol”. 103 (2), s. 217–222, 2012. DOI: 10.1016/j.radonc.2012.01.007. PMID: 22341794.
- ↑ B.R. Pieters, D.Z. de Back, C.C. Koning, A.H. Zwinderman. Comparison of three radiotherapy modalities on biochemical control and overall survival for the treatment of prostate cancer: a systematic review. „Radiother Oncol”. 93 (2), s. 168–173, 2009. DOI: 10.1016/j.radonc.2009.08.033. PMID: 19748692.
- ↑ Mottet i in. 2017 ↓, s. 46–47.
- ↑ Gunderson i Tepper 2012 ↓, s. 1069–1070.
- ↑ DeVita, Lawrence i Rosenberg 2015 ↓, s. 957.
- ↑ M. Bolla, G. Van Tienhoven, P. Warde, J.B. Dubois i inni. External irradiation with or without long-term androgen suppression for prostate cancer with high metastatic risk: 10-year results of an EORTC randomised study. „Lancet Oncol”. 11 (11), s. 1066–1073, 2010. DOI: 10.1016/S1470-2045(10)70223-0. PMID: 20933466.
- ↑ M.V. Pilepich, K. Winter, C.A. Lawton, R.E. Krisch i inni. Androgen suppression adjuvant to definitive radiotherapy in prostate carcinoma--long-term results of phase III RTOG 85-31. „Int J Radiat Oncol Biol Phys”. 61 (5), s. 1285–1290, 2005. DOI: 10.1016/j.ijrobp.2004.08.047. PMID: 15817329.
- ↑ M.D. Mason, W.R. Parulekar, M.R. Sydes, M. Brundage i inni. Final Report of the Intergroup Randomized Study of Combined Androgen-Deprivation Therapy Plus Radiotherapy Versus Androgen-Deprivation Therapy Alone in Locally Advanced Prostate Cancer. „J Clin Oncol”. 33 (19), s. 2143–2150, 2015. DOI: 10.1200/JCO.2014.57.7510. PMID: 25691677.
- ↑ P. Warde, M. Mason, K. Ding, P. Kirkbride i inni. Combined androgen deprivation therapy and radiation therapy for locally advanced prostate cancer: a randomised, phase 3 trial. „Lancet”. 378 (9809), s. 2104–2111, 2011. DOI: 10.1016/S0140-6736(11)61095-7. PMID: 22056152.
- ↑ A. Widmark, O. Klepp, A. Solberg, J.E. Damber i inni. Endocrine treatment, with or without radiotherapy, in locally advanced prostate cancer (SPCG-7/SFUO-3): an open randomised phase III trial. „Lancet”. 373 (9660), s. 301–308, 2009. DOI: 10.1016/S0140-6736(08)61815-2. PMID: 19091394.
- ↑ S.D. Fosså, F. Wiklund, O. Klepp, A. Angelsen i inni. Ten- and 15-yr Prostate Cancer-specific Mortality in Patients with Nonmetastatic Locally Advanced or Aggressive Intermediate Prostate Cancer, Randomized to Lifelong Endocrine Treatment Alone or Combined with Radiotherapy: Final Results of The Scandinavian Prostate Cancer Group-7. „Eur Urol”. 70 (4), s. 684–691, 2016. DOI: 10.1016/j.eururo.2016.03.021. PMID: 27025586.
- ↑ Mohler i in. 2017 ↓, s. 63.
- ↑ J.W. Denham, A. Steigler, D.S. Lamb, D. Joseph i inni. Short-term neoadjuvant androgen deprivation and radiotherapy for locally advanced prostate cancer: 10-year data from the TROG 96.01 randomised trial. „Lancet Oncol”. 12 (5), s. 451–459, 2011. DOI: 10.1016/S1470-2045(11)70063-8. PMID: 21440505.
- ↑ C.U. Jones, D. Hunt, D.G. McGowan, M.B. Amin i inni. Radiotherapy and short-term androgen deprivation for localized prostate cancer. „N Engl J Med”. 365 (2), s. 107–118, 2011. DOI: 10.1056/NEJMoa1012348. PMID: 21751904.
- ↑ a b W.U. Shipley, W. Seiferheld, H.R. Lukka, P.P. Major i inni. Radiation with or without Antiandrogen Therapy in Recurrent Prostate Cancer. „N Engl J Med”. 376 (5), s. 417–428, 2017. DOI: 10.1056/NEJMoa1607529. PMID: 28146658.
- ↑ A.V. D’Amico, M.H. Chen, A.A. Renshaw, M. Loffredo i inni. Androgen suppression and radiation vs radiation alone for prostate cancer: a randomized trial. „JAMA”. 299 (3), s. 289–295, 2008. DOI: 10.1001/jama.299.3.289. PMID: 18212313.
- ↑ a b Mohler i in. 2017 ↓, s. 62.
- ↑ T.M. Pisansky, D. Hunt, L.G. Gomella, M.B. Amin i inni. Duration of androgen suppression before radiotherapy for localized prostate cancer: radiation therapy oncology group randomized clinical trial 9910. „J Clin Oncol”. 33 (4), s. 332–339, 2015. DOI: 10.1200/JCO.2014.58.0662. PMID: 25534388.
- ↑ G.L. Lu-Yao, P.C. Albertsen, D.F. Moore, W. Shih i inni. Fifteen-year survival outcomes following primary androgen-deprivation therapy for localized prostate cancer. „JAMA Intern Med”. 174 (9), s. 1460–1467, 2014. DOI: 10.1001/jamainternmed.2014.3028. PMID: 25023796.
- ↑ A.L. Potosky, R. Haque, A.E. Cassidy-Bushrow, M. Ulcickas Yood i inni. Effectiveness of primary androgen-deprivation therapy for clinically localized prostate cancer. „J Clin Oncol”. 32 (13), s. 1324–1330, 2014. DOI: 10.1200/JCO.2013.52.5782. PMID: 24638009.
- ↑ a b c d Rutkowski i Warzocha 2013 ↓, s. 346.
- ↑ DeVita, Lawrence i Rosenberg 2015 ↓, s. 963.
- ↑ a b DeVita, Lawrence i Rosenberg 2015 ↓, s. 963–964.
- ↑ P. Pronzato, M. Rondini. Hormonotherapy of advanced prostate cancer. „Ann Oncol”. 16 Suppl 4, s. 80–84, 2005. DOI: 10.1093/annonc/mdi913. PMID: 15923436.
- ↑ Albala i in. 2011 ↓, s. 234.
- ↑ a b L.A. Kluth, S.F. Shariat, C. Kratzik, S. Tagawa i inni. The hypothalamic-pituitary-gonadal axis and prostate cancer: implications for androgen deprivation therapy. „World J Urol”. 32 (3), s. 669–676, 2014. DOI: 10.1007/s00345-013-1157-5. PMID: 23999854.
- ↑ M. Kovacs, A.V. Schally. Comparison of mechanisms of action of luteinizing hormone-releasing hormone (LHRH) antagonist cetrorelix and LHRH agonist triptorelin on the gene expression of pituitary LHRH receptors in rats. „Proc Natl Acad Sci U S A”. 98 (21), s. 12197–12202, 2001. DOI: 10.1073/pnas.211442598. PMID: 11593037.
- ↑ B. Dybowski. Degareliks – antagonista LHRH w terapii zaawansowanego raka stercza. Przegl Urolog 2011/5 (69).
- ↑ a b c F. Ricci, G. Buzzatti, A. Rubagotti, F. Boccardo. Safety of antiandrogen therapy for treating prostate cancer. „Expert Opin Drug Saf”. 13 (11), s. 1483–1499, 2014. DOI: 10.1517/14740338.2014.966686. PMID: 25270521.
- ↑ Mohler i in. 2017 ↓, s. 16.
- ↑ a b c d e f g Mottet i in. 2017 ↓, s. 57.
- ↑ a b c d Krzakowski i in. 2015 ↓, s. 748.
- ↑ a b c d H. Habchi, N. Mottet. Androgen Deprivation Therapy in Prostate Cancer – Current Status in M1 Patients. „Oncol Res Treat”. 38 (12), s. 646–652, 2015. DOI: 10.1159/000441734. PMID: 26633005.
- ↑ F. Schröder, E.D. Crawford, K. Axcrona, H. Payne i inni. Androgen deprivation therapy: past, present and future. „BJU Int”. 109 Suppl 6, s. 1–12, 2012. DOI: 10.1111/j.1464-410X.2012.11215.x. PMID: 22672120.
- ↑ a b c Mottet i in. 2017 ↓, s. 55.
- ↑ B. Nair, T. Wilt, R. MacDonald, I. Rutks. Early versus deferred androgen suppression in the treatment of advanced prostatic cancer. „Cochrane Database Syst Rev”, 2002. DOI: 10.1002/14651858.CD003506. PMID: 11869665.
- ↑ DeVita, Lawrence i Rosenberg 2015 ↓, s. 968.
- ↑ Krzakowski i in. 2015 ↓, s. 747–748.
- ↑ a b c Mottet i in. 2017 ↓, s. 53.
- ↑ a b DeVita, Lawrence i Rosenberg 2015 ↓, s. 966.
- ↑ F. Kunath, H.R. Grobe, G. Rücker, E. Motschall i inni. Non-steroidal antiandrogen monotherapy compared with luteinising hormone-releasing hormone agonists or surgical castration monotherapy for advanced prostate cancer. „Cochrane Database Syst Rev”, 2014. DOI: 10.1002/14651858.CD009266.pub2. PMID: 24979481.
- ↑ a b c d e f g Mottet i in. 2017 ↓, s. 54.
- ↑ M.A. Eisenberger, B.A. Blumenstein, E.D. Crawford, G. Miller i inni. Bilateral orchiectomy with or without flutamide for metastatic prostate cancer. „N Engl J Med”. 339 (15), s. 1036–1042, 1998. DOI: 10.1056/NEJM199810083391504. PMID: 9761805.
- ↑ Maximum androgen blockade in advanced prostate cancer: an overview of the randomised trials. Prostate Cancer Trialists Collaborative Group. „Lancet”. 355 (9214), s. 1491–1498, 2000. PMID: 10801170.
- ↑ B. Schmitt, C. Bennett, J. Seidenfeld, D. Samson i inni. Maximal androgen blockade for advanced prostate cancer. „Cochrane Database Syst Rev”, 2000. DOI: 10.1002/14651858.CD001526. PMID: 10796804.
- ↑ H. Akaza, S. Hinotsu, M. Usami, Y. Arai i inni. Combined androgen blockade with bicalutamide for advanced prostate cancer: long-term follow-up of a phase 3, double-blind, randomized study for survival. „Cancer”. 115 (15), s. 3437–3445, 2009. DOI: 10.1002/cncr.24395. PMID: 19536889.
- ↑ Mottet i in. 2017 ↓, s. 55–56.
- ↑ G. Gravis, K. Fizazi, F. Joly, S. Oudard i inni. Androgen-deprivation therapy alone or with docetaxel in non-castrate metastatic prostate cancer (GETUG-AFU 15): a randomised, open-label, phase 3 trial. „Lancet Oncol”. 14 (2), s. 149–158, 2013. DOI: 10.1016/S1470-2045(12)70560-0. PMID: 23306100.
- ↑ G. Gravis, J.M. Boher, F. Joly, M. Soulié i inni. Androgen Deprivation Therapy (ADT) Plus Docetaxel Versus ADT Alone in Metastatic Non castrate Prostate Cancer: Impact of Metastatic Burden and Long-term Survival Analysis of the Randomized Phase 3 GETUG-AFU15 Trial. „Eur Urol”. 70 (2), s. 256–262, 2016. DOI: 10.1016/j.eururo.2015.11.005. PMID: 26610858.
- ↑ C.J. Sweeney, Y.H. Chen, M. Carducci, G. Liu i inni. Chemohormonal Therapy in Metastatic Hormone-Sensitive Prostate Cancer. „N Engl J Med”. 373 (8), s. 737–746, 2015. DOI: 10.1056/NEJMoa1503747. PMID: 26244877.
- ↑ N.D. James, M.R. Sydes, N.W. Clarke, M.D. Mason i inni. Addition of docetaxel, zoledronic acid, or both to first-line long-term hormone therapy in prostate cancer (STAMPEDE): survival results from an adaptive, multiarm, multistage, platform randomised controlled trial. „Lancet”. 387 (10024), s. 1163–1177, 2016. DOI: 10.1016/S0140-6736(15)01037-5. PMID: 26719232.
- ↑ Mottet i in. 2017 ↓, s. 54–56.
- ↑ M. Hussain, C.M. Tangen, D.L. Berry, C.S. Higano i inni. Intermittent versus continuous androgen deprivation in prostate cancer. „N Engl J Med”. 368 (14), s. 1314–1325, 2013. DOI: 10.1056/NEJMoa1212299. PMID: 23550669.
- ↑ D. Brungs, J. Chen, P. Masson, R.J. Epstein. Intermittent androgen deprivation is a rational standard-of-care treatment for all stages of progressive prostate cancer: results from a systematic review and meta-analysis. „Prostate Cancer Prostatic Dis”. 17 (2), s. 105–111, 2014. DOI: 10.1038/pcan.2014.10. PMID: 24686773.
- ↑ S. Magnan, R. Zarychanski, L. Pilote, L. Bernier i inni. Intermittent vs Continuous Androgen Deprivation Therapy for Prostate Cancer: A Systematic Review and Meta-analysis. „JAMA Oncol”. 1 (9), s. 1261–1269, 2015. DOI: 10.1001/jamaoncol.2015.2895. PMID: 26378418.
- ↑ M. Hussain, C. Tangen, C. Higano, N. Vogelzang i inni. Evaluating Intermittent Androgen-Deprivation Therapy Phase III Clinical Trials: The Devil Is in the Details. „J Clin Oncol”. 34 (3), s. 280–285, 2016. DOI: 10.1200/JCO.2015.62.8065. PMID: 26552421.
- ↑ Michał Jakubczyk, Maciej Niewada: Elementy oceny organizacji i wyników badań klinicznych. Centrum Medyczne Kształcenia Podyplomowego, 2011, s. 32–33. ISBN 978-83-62110-29-2.
- ↑ DeVita, Lawrence i Rosenberg 2015 ↓, s. 960.
- ↑ C.J. Paller, E.S. Antonarakis. Management of biochemically recurrent prostate cancer after local therapy: evolving standards of care and new directions. „Clin Adv Hematol Oncol”. 11 (1), s. 14–23, 2013. PMID: 23416859.
- ↑ a b c Mohler i in. 2017 ↓, s. 77.
- ↑ a b Mottet i in. 2017 ↓, s. 70.
- ↑ a b Mottet i in. 2017 ↓, s. 73.
- ↑ Mottet i in. 2017 ↓, s. 71.
- ↑ B.J. Trock, M. Han, S.J. Freedland, E.B. Humphreys i inni. Prostate cancer-specific survival following salvage radiotherapy vs observation in men with biochemical recurrence after radical prostatectomy. „JAMA”. 299 (23), s. 2760–2769, 2008. DOI: 10.1001/jama.299.23.2760. PMID: 18560003.
- ↑ S.E. Cotter, M.H. Chen, J.W. Moul, W.R. Lee i inni. Salvage radiation in men after prostate-specific antigen failure and the risk of death. „Cancer”. 117 (17), s. 3925–3932, 2011. DOI: 10.1002/cncr.25993. PMID: 21437885.
- ↑ a b c Mohler i in. 2017 ↓, s. 78.
- ↑ C. Carrie, A. Hasbini, G. de Laroche, P. Richaud i inni. Salvage radiotherapy with or without short-term hormone therapy for rising prostate-specific antigen concentration after radical prostatectomy (GETUG-AFU 16): a randomised, multicentre, open-label phase 3 trial. „Lancet Oncol”. 17 (6), s. 747–756, 2016. DOI: 10.1016/S1470-2045(16)00111-X. PMID: 27160475.
- ↑ a b c Mottet i in. 2017 ↓, s. 76.
- ↑ G.M. Duchesne, H.H. Woo, J.K. Bassett, S.J. Bowe i inni. Timing of androgen-deprivation therapy in patients with prostate cancer with a rising PSA (TROG 03.06 and VCOG PR 01-03 [TOAD]): a randomised, multicentre, non-blinded, phase 3 trial. „Lancet Oncol”. 17 (6), s. 727–737, 2016. DOI: 10.1016/S1470-2045(16)00107-8. PMID: 27155740.
- ↑ R.C. van den Bergh, N.J. van Casteren, T. van den Broeck, E.R. Fordyce i inni. Role of Hormonal Treatment in Prostate Cancer Patients with Nonmetastatic Disease Recurrence After Local Curative Treatment: A Systematic Review. „Eur Urol”. 69 (5), s. 802–820, 2016. DOI: 10.1016/j.eururo.2015.11.023. PMID: 26691493.
- ↑ a b Mottet i in. 2017 ↓, s. 72.
- ↑ G.T. Gotto, L.H. Yunis, K. Vora, J.A. Eastham i inni. Impact of prior prostate radiation on complications after radical prostatectomy. „J Urol”. 184 (1), s. 136–142, 2010. DOI: 10.1016/j.juro.2010.03.031. PMID: 20478594.
- ↑ A.J. Stephenson, P.T. Scardino, F.J. Bianco, C.J. DiBlasio i inni. Morbidity and functional outcomes of salvage radical prostatectomy for locally recurrent prostate cancer after radiation therapy. „J Urol”. 172, s. 2239–2243, 2004. PMID: 15538239.
- ↑ a b D.C. Chade, J. Eastham, M. Graefen, J.C. Hu i inni. Cancer control and functional outcomes of salvage radical prostatectomy for radiation-recurrent prostate cancer: a systematic review of the literature. „Eur Urol”. 61 (5), s. 961–971, 2012. DOI: 10.1016/j.eururo.2012.01.022. PMID: 22280856.
- ↑ Mottet i in. 2017 ↓, s. 74.
- ↑ a b Mottet i in. 2017 ↓, s. 75.
- ↑ C.P. Chen, V. Weinberg, K. Shinohara, M. Roach i inni. Salvage HDR brachytherapy for recurrent prostate cancer after previous definitive radiation therapy: 5-year outcomes. „Int J Radiat Oncol Biol Phys”. 86 (2), s. 324–329, 2013. DOI: 10.1016/j.ijrobp.2013.01.027. PMID: 23474112.
- ↑ R.J. Burri, N.N. Stone, P. Unger, R.G. Stock. Long-term outcome and toxicity of salvage brachytherapy for local failure after initial radiotherapy for prostate cancer. „Int J Radiat Oncol Biol Phys”. 77 (5), s. 1338–1344, 2010. DOI: 10.1016/j.ijrobp.2009.06.061. PMID: 20138442.
- ↑ Y. Yamada, M.A. Kollmeier, X. Pei, C.C. Kan i inni. A Phase II study of salvage high-dose-rate brachytherapy for the treatment of locally recurrent prostate cancer after definitive external beam radiotherapy. „Brachytherapy”. 13 (2). s. 111–116. DOI: 10.1016/j.brachy.2013.11.005. PMID: 24373762.
- ↑ B. Lee, K. Shinohara, V. Weinberg, A.R. Gottschalk i inni. Feasibility of high-dose-rate brachytherapy salvage for local prostate cancer recurrence after radiotherapy: the University of California-San Francisco experience. „Int J Radiat Oncol Biol Phys”. 67 (4), s. 1106–1112, 2007. DOI: 10.1016/j.ijrobp.2006.10.012. PMID: 17197119.
- ↑ a b Mottet i in. 2017 ↓, s. 74–75.
- ↑ M. Ismail, S. Ahmed, C. Kastner, J. Davies. Salvage cryotherapy for recurrent prostate cancer after radiation failure: a prospective case series of the first 100 patients. „BJU Int”. 100 (4), s. 760–764, 2007. DOI: 10.1111/j.1464-410X.2007.07045.x. PMID: 17662081.
- ↑ L.L. Pisters, J.C. Rewcastle, B.J. Donnelly, F.M. Lugnani i inni. Salvage prostate cryoablation: initial results from the cryo on-line data registry. „J Urol”. 180 (2), s. 559–563, 2008. DOI: 10.1016/j.juro.2008.04.005. PMID: 18554664.
- ↑ a b L.L. Pisters, D. Leibovici, M. Blute, H. Zincke i inni. Locally recurrent prostate cancer after initial radiation therapy: a comparison of salvage radical prostatectomy versus cryotherapy. „J Urol”. 182 (2), s. 517–525, 2009. DOI: 10.1016/j.juro.2009.04.006. PMID: 19524984.
- ↑ Mottet i in. 2017 ↓, s. 75–76.
- ↑ a b c DeVita, Lawrence i Rosenberg 2015 ↓, s. 970.
- ↑ a b c d Mottet i in. 2017 ↓, s. 77.
- ↑ S.J. Hotte, F. Saad. Current management of castrate-resistant prostate cancer. „Curr Oncol”. 17 Suppl 2, 2010. PMID: 20882137.
- ↑ a b c Mottet i in. 2017 ↓, s. 83.
- ↑ Sławomir Poletajew. Opcje terapeutyczne u pacjentów z opornym na kastrację rakiem stercza z przerzutami. „Przegląd Urologiczny”, 2016.
- ↑ a b M. Poorthuis, R. Vernooij, R. van Moorselaar, T.M. de Reijke. Second-line therapy in patients with metastatic castration-resistant prostate cancer with progression after or under docetaxel: A systematic review of nine randomized controlled trials. „Semin Oncol”. 44 (5), s. 358–371, 2017. DOI: 10.1053/j.seminoncol.2017.10.005. PMID: 29580437.
- ↑ a b c D. Lorente, K. Fizazi, C. Sweeney, J.S. de Bono. Optimal Treatment Sequence for Metastatic Castration-resistant Prostate Cancer. „Eur Urol Focus”. 2 (5), s. 488–498, 2016. DOI: 10.1016/j.euf.2016.10.008. PMID: 28723514.
- ↑ a b c d Mottet i in. 2017 ↓, s. 82.
- ↑ a b c d e f Mohler i in. 2017 ↓, s. 82.
- ↑ M. Hussain, M. Wolf, E. Marshall, E.D. Crawford i inni. Effects of continued androgen-deprivation therapy and other prognostic factors on response and survival in phase II chemotherapy trials for hormone-refractory prostate cancer: a Southwest Oncology Group report. „J Clin Oncol”. 12 (9), s. 1868–1875, 1994. DOI: 10.1200/JCO.1994.12.9.1868. PMID: 8083710.
- ↑ C.D. Taylor, P. Elson, D.L. Trump. Importance of continued testicular suppression in hormone-refractory prostate cancer. „J Clin Oncol”. 11 (11), s. 2167–2172, 1993. DOI: 10.1200/JCO.1993.11.11.2167. PMID: 8229130.
- ↑ Y. Rehman, J.E. Rosenberg. Abiraterone acetate: oral androgen biosynthesis inhibitor for treatment of castration-resistant prostate cancer. „Drug Des Devel Ther”. 6, s. 13–18, 2012. DOI: 10.2147/DDDT.S15850. PMID: 22291466.
- ↑ B.A. Gartrell, F. Saad. Abiraterone in the management of castration-resistant prostate cancer prior to chemotherapy. „Ther Adv Urol”. 7 (4), s. 194–202, 2015. DOI: 10.1177/1756287215592288. PMID: 26445599.
- ↑ a b Mohler i in. 2017 ↓, s. 68.
- ↑ Mottet i in. 2017 ↓, s. 78.
- ↑ C.J. Ryan, M.R. Smith, J.S. de Bono, A. Molina i inni. Abiraterone in metastatic prostate cancer without previous chemotherapy. „N Engl J Med”. 368 (2), s. 138–148, 2013. DOI: 10.1056/NEJMoa1209096. PMID: 23228172.
- ↑ C.J. Ryan, M.R. Smith, K. Fizazi, F. Saad i inni. Abiraterone acetate plus prednisone versus placebo plus prednisone in chemotherapy-naive men with metastatic castration-resistant prostate cancer (COU-AA-302): final overall survival analysis of a randomised, double-blind, placebo-controlled phase 3 study. „Lancet Oncol”. 16 (2), s. 152–160, 2015. DOI: 10.1016/S1470-2045(14)71205-7. PMID: 25601341.
- ↑ Stein CA, Levin R, Given R, et al. Randomized phase 2 therapeutic equivalence study of abiraterone acetate fine particle formulation vs. originator abiraterone acetate in patients with metastatic castration-resistant prostate cancer: The STAAR study. Urol Oncol. 2018;36(2):81e9-81.e16.
- ↑ J. Schalken, J.M. Fitzpatrick. Enzalutamide: targeting the androgen signalling pathway in metastatic castration-resistant prostate cancer. „BJU Int”. 117 (2), s. 215–225, 2016. DOI: 10.1111/bju.13123. PMID: 25818596.
- ↑ C.P. Evans, C.S. Higano, T. Keane, G. Andriole i inni. The PREVAIL Study: Primary Outcomes by Site and Extent of Baseline Disease for Enzalutamide-treated Men with Chemotherapy-naïve Metastatic Castration-resistant Prostate Cancer. „Eur Urol”. 70 (4), s. 675–683, 2016. DOI: 10.1016/j.eururo.2016.03.017. PMID: 27006332.
- ↑ J.N. Graff, E.D. Chamberlain. Sipuleucel-T in the treatment of prostate cancer: an evidence-based review of its place in therapy. „Core Evid”. 10, s. 1–10, 2015. DOI: 10.2147/CE.S54712. PMID: 25565923.
- ↑ C.E. Handy, E.S. Antonarakis. Sipuleucel-T for the treatment of prostate cancer: novel insights and future directions. „Future Oncol”. 14 (10), s. 907–917, 2018. DOI: 10.2217/fon-2017-0531. PMID: 29260582.
- ↑ a b DeVita, Lawrence i Rosenberg 2015 ↓, s. 972.
- ↑ a b Mohler i in. 2017 ↓, s. 81.
- ↑ a b c Mohler i in. 2017 ↓, s. 34.
- ↑ P.W. Kantoff, C.S. Higano, N.D. Shore, E.R. Berger i inni. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. „N Engl J Med”. 363 (5), s. 411–422, 2010. DOI: 10.1056/NEJMoa1001294. PMID: 20818862.
- ↑ a b I.F. Tannock, R. de Wit, W.R. Berry, J. Horti i inni. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. „N Engl J Med”. 351 (15), s. 1502–1512, 2004. DOI: 10.1056/NEJMoa040720. PMID: 15470213.
- ↑ D.R. Berthold, G.R. Pond, F. Soban, R. de Wit i inni. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer: updated survival in the TAX 327 study. „J Clin Oncol”. 26 (2), s. 242–245, 2008. DOI: 10.1200/JCO.2007.12.4008. PMID: 18182665.
- ↑ a b Mohler i in. 2017 ↓, s. 70.
- ↑ H.I. Scher, S. Halabi, I. Tannock, M. Morris i inni. Design and end points of clinical trials for patients with progressive prostate cancer and castrate levels of testosterone: recommendations of the Prostate Cancer Clinical Trials Working Group. „J Clin Oncol”. 26 (7), s. 1148–1159, 2008. DOI: 10.1200/JCO.2007.12.4487. PMID: 18309951.
- ↑ DeVita, Lawrence i Rosenberg 2015 ↓, s. 971.
- ↑ DeVita, Lawrence i Rosenberg 2015 ↓, s. 973.
- ↑ a b F. Zustovich, D. Pastorelli. Therapeutic management of bone metastasis in prostate cancer: an update. „Expert Rev Anticancer Ther”, s. 1–13, 2016. DOI: 10.1080/14737140.2016.1241148. PMID: 27666299.
- ↑ Mottet i in. 2017 ↓, s. 81–82.
- ↑ S.M. Dy, S.M. Asch, A. Naeim, H. Sanati i inni. Evidence-based standards for cancer pain management. „J Clin Oncol”. 26 (23), s. 3879–3885, 2008. DOI: 10.1200/JCO.2007.15.9517. PMID: 18688056.
- ↑ a b F. Macedo, K. Ladeira, F. Pinho, N. Saraiva i inni. Bone Metastases: An Overview. „Oncol Rev”. 11 (1), s. 321, 2017. DOI: 10.4081/oncol.2017.321. PMID: 28584570.
- ↑ DeVita, Lawrence i Rosenberg 2015 ↓, s. 973–974.
- ↑ a b DeVita, Lawrence i Rosenberg 2015 ↓, s. 974.
- ↑ a b c d Mottet i in. 2017 ↓, s. 80.
- ↑ C. Parker, S. Nilsson, D. Heinrich, S.I. Helle i inni. Alpha emitter radium-223 and survival in metastatic prostate cancer. „N Engl J Med”. 369 (3), s. 213–223, 2013. DOI: 10.1056/NEJMoa1213755. PMID: 23863050.
- ↑ P. Hoskin, O. Sartor, J.M. O’Sullivan, D.C. Johannessen i inni. Efficacy and safety of radium-223 dichloride in patients with castration-resistant prostate cancer and symptomatic bone metastases, with or without previous docetaxel use: a prespecified subgroup analysis from the randomised, double-blind, phase 3 ALSYMPCA trial. „Lancet Oncol”. 15 (12), s. 1397–1406, 2014. DOI: 10.1016/S1470-2045(14)70474-7. PMID: 25439694.
- ↑ Mottet i in. 2017 ↓, s. 80–81.
- ↑ M.R. Smith, F. Saad, R. Coleman, N. Shore i inni. Denosumab and bone-metastasis-free survival in men with castration-resistant prostate cancer: results of a phase 3, randomised, placebo-controlled trial. „Lancet”. 379 (9810), s. 39–46, 2012. DOI: 10.1016/S0140-6736(11)61226-9. PMID: 22093187.
- ↑ a b Mohler i in. 2017 ↓, s. 19.
- ↑ a b Mohler i in. 2017 ↓, s. 83.
- ↑ J.S. de Bono, S. Oudard, M. Ozguroglu, S. Hansen i inni. Prednisone plus cabazitaxel or mitoxantrone for metastatic castration-resistant prostate cancer progressing after docetaxel treatment: a randomised open-label trial. „Lancet”. 376 (9747), s. 1147–1154, 2010. DOI: 10.1016/S0140-6736(10)61389-X. PMID: 20888992.
- ↑ S. Oudard, K. Fizazi, L. Sengeløv, G. Daugaard i inni. Cabazitaxel Versus Docetaxel As First-Line Therapy for Patients With Metastatic Castration-Resistant Prostate Cancer: A Randomized Phase III Trial-FIRSTANA. „J Clin Oncol”. 35 (28), s. 3189–3197, 2017. DOI: 10.1200/JCO.2016.72.1068. PMID: 28753384.
- ↑ J.S. de Bono, C.J. Logothetis, A. Molina, K. Fizazi i inni. Abiraterone and increased survival in metastatic prostate cancer. „N Engl J Med”. 364 (21), s. 1995–2005, 2011. DOI: 10.1056/NEJMoa1014618. PMID: 21612468.
- ↑ K. Fizazi, H.I. Scher, A. Molina, C.J. Logothetis i inni. Abiraterone acetate for treatment of metastatic castration-resistant prostate cancer: final overall survival analysis of the COU-AA-301 randomised, double-blind, placebo-controlled phase 3 study. „Lancet Oncol”. 13 (10), s. 983–992, 2012. DOI: 10.1016/S1470-2045(12)70379-0. PMID: 22995653.
- ↑ H.I. Scher, K. Fizazi, F. Saad, M.E. Taplin i inni. Increased survival with enzalutamide in prostate cancer after chemotherapy. „N Engl J Med”. 367 (13), s. 1187–1197, 2012. DOI: 10.1056/NEJMoa1207506. PMID: 22894553.
- ↑ Mohler i in. 2017 ↓, s. 84.
- ↑ N Howlader i inni, Contents of the SEER Cancer Statistics Review (CSR), 1975-2014 Table 23.8 Cancer of the Prostate (Invasive) [zarchiwizowane z adresu 2018-07-28].
- ↑ a b cancer.gov: Review: summary staging. [dostęp 2019-10-30]. [zarchiwizowane z tego adresu (2017-10-30)].
- ↑ Donald J. Meuten: Tumors in Domestic Animals. John Wiley & Sons, 2008, s. 568–570. ISBN 978-0-470-37670-6.
- ↑ a b c Stephen J. Withrow: Withrow and MacEwen’s Small Animal Clinical Oncology. Elsevier Health Sciences, 2007, s. 641–643. ISBN 978-0-7216-0558-6.
- ↑ a b c d Stephen J. Ettinger, Edward C. Feldman: Textbook of Veterinary Internal Medicine. Elsevier Health Science, 2009, s. 2209–2210. ISBN 978-1-4377-0282-8.
- ↑ a b c d e f g h i j k l m n S.R. Denmeade, J.T. Isaacs. A history of prostate cancer treatment. „Nat Rev Cancer”. 2 (5), s. 389–396, 2002. DOI: 10.1038/nrc801. PMID: 12044015.
- ↑ K. Ghabili, J.J. Tosoian, E.M. Schaeffer, C.P. Pavlovich i inni. The History of Prostate Cancer From Antiquity: Review of Paleopathological Studies. „Urology”. 97, s. 8–12, 2016. DOI: 10.1016/j.urology.2016.08.032. PMID: 27591810.
- ↑ a b c d e S. Sriprasad, M.R. Feneley, P.M. Thompson. History of prostate cancer treatment. „Surg Oncol”. 18 (3), s. 185–191, 2009. DOI: 10.1016/j.suronc.2009.07.001. PMID: 19647427.
- ↑ J.A. Vilchez-Martinez, E. Pedroza, A. Arimura, A.V. Schally. Paradoxical effects of D-Trp6-luteinizing hormone-releasing hormone on the hypothalamic-pituitary-gonadal axis in immature female rats. „Fertil Steril”. 31 (6), s. 677–682, 1979. PMID: 376360.
- ↑ J. Sandow, W. Von Rechenberg, G. Jerzabek, W. Stoll. Pituitary gonadotropin inhibition by a highly active analog of luteinizing hormone-releasing hormone. „Fertil Steril”. 30 (2), s. 205–209, 1978. PMID: 354980.
- ↑ R.B. Smith, P.C. Walsh, W.E. Goodwin. Cyproterone acetate in the treatment of advanced carcinoma of the prostate. „J Urol”. 110 (1), s. 106–108, 1973. PMID: 4713345.
- ↑ W.W. Scott, D.E. Johnson, J.E. Schmidt, R.P. Gibbons i inni. Chemotherapy of advanced prostatic carcinoma with cyclophosphamide or 5-fluorouracil: results of first national randomized study. „J Urol”. 114 (6), s. 909–911, 1975. PMID: 1104900.
- ↑ I.F. Tannock, D. Osoba, M.R. Stockler, D.S. Ernst i inni. Chemotherapy with mitoxantrone plus prednisone or prednisone alone for symptomatic hormone-resistant prostate cancer: a Canadian randomized trial with palliative end points. „J Clin Oncol”. 14 (6), s. 1756–1764, 1996. DOI: 10.1200/JCO.1996.14.6.1756. PMID: 8656243.
- ↑ P.W. Kantoff, S. Halabi, M. Conaway, J. Picus i inni. Hydrocortisone with or without mitoxantrone in men with hormone-refractory prostate cancer: results of the cancer and leukemia group B 9182 study. „J Clin Oncol”. 17 (8), s. 2506–2513, 1999. DOI: 10.1200/JCO.1999.17.8.2506. PMID: 10561316.
Bibliografia
- N. Mottet, J Bellmunt, E. Briers, M. Bolla i inni. EAU-ESTRO-ESUR-SIOG Guidelines on Prostate Cancer. „European Urology”. 71 (4), s. 618–629, 2017. DOI: 10.1016/j.eururo.2016.08.003. ISSN 0302-2838.
- J.L. Mohler, A.J. Armstrong, R.R. Bahnson, A.V. D’Amico i inni. Prostate Cancer, Version 2.2017.. „Journal of the National Comprehensive Cancer Network”, s. 804–834, 2017. DOI: 10.6004/jnccn.2017.0100. ISSN 1540-1405.
- C. Parker, S. Gillessen, A. Heidenreich, A. Horwich i inni. Cancer of the prostate: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. „Annals of Oncology”. 26 Suppl 5, 2015. DOI: 10.1093/annonc/mdv222. ISSN 0923-7534. PMID: 26205393.
- Maciej Krzakowski, Piotr Potemski, Krzysztof Warzocha, Pior Wysocki: Onkologia kliniczna. T. II. Gdańsk: Via Medica, 2015. ISBN 978-83-7599-796-5.
- John N. Eble, Guido Sauter, Jonathan I. Epstein, Isabell A. Sesterhenn: Pathology and Genetics of Tumours of the Urinary System and Male Genital Organs. IARC Press, 2004.
- Vincent T. DeVita, Theodore S. Lawrence, Steven A. Rosenberg: DeVita, Hellman, and Rosenberg’s Cancer Principles & Practice of Oncology. Wolters Kluwer Health, 2015. ISBN 978-1-4511-9294-0.
- Jerzy Stachura, Domagała Wenancjusz: Patologia znaczy słowo o chorobie. T. II. Polska Akademia Umiejętności, 2009. ISBN 978-83-60183-64-9.
- Christopher D.M. Fletcher: Diagnostic Histopathology of Tumors. Elsevier Health Sciences, 2013. ISBN 978-1-4557-3754-3.
- Piotr Rutkowski, Krzysztof Warzocha: Zalecenia postępowania diagnostyczno-terapeutycznego w nowotworach złośliwych 2013 rok. VM Media, 2013. ISBN 978-83-7599-594-7.
- Leonard Gunderson, Joel Tepper: Clinical Radiation Oncology 3rd Edition. Elsevier Inc, 2012. ISBN 978-1-4377-1637-5.
- David M. Albala, Allen F. Morey, Leonard G. Gomella, John P. Stein: Oxford American Handbook of Urology. Oxford University Press, 2011. ISBN 978-0-19-537139-0.
- John Blandy, Amir Kaisary: Urology. Wiley-Blackwell, 2009. ISBN 978-1-4051-2270-2.
![]() Przeczytaj ostrzeżenie dotyczące informacji medycznych i pokrewnych zamieszczonych w Wikipedii.
Przeczytaj ostrzeżenie dotyczące informacji medycznych i pokrewnych zamieszczonych w Wikipedii.
Media użyte na tej stronie

The Star of Life, medical symbol used on some ambulances.
Star of Life was designed/created by a National Highway Traffic Safety Administration (US Gov) employee and is thus in the public domain.Autor: Oryginalnym przesyłającym był M502114 z angielskiej Wikipedii, Licencja: CC-BY-SA-3.0
Zoladex (goserelin) 3.6mg & 10.8mg
Autor: Yale Rosen from USA, Licencja: CC BY-SA 2.0
Bilateral nodules of metastatic carcinoma
Autor: Yale Rosen from USA, Licencja: CC BY-SA 2.0
Metastatic prostatic adenocarcinoma
Autor: Nephron, Licencja: CC BY-SA 3.0
Micrograph of prostate adenocarcinoma. Needle core biopsy. HPS stain.
See also
- Image:Perineural invasion prostate high mag.jpg - same case, view demonstrating perineural invasion.
- Image:Prostate adenocarcinoma 2 high mag hps.jpg - same case, different core showing benign (normal) prostatic glands and malignant ones - high magnification.
- Image:Prostate adenocarcinoma 2 intermed mag hps.jpg - same case, different core showing benign (normal) prostatic glands and malignant ones - intermediate magnification.
Autor:
- InvictaHOG z angielskiej Wikipedii
- Na Commons przeniósł z en.wikipedia użytkownik Leptictidium z pomocą narzędzia CommonsHelper.
- derivative work: M1llx (zgłoś błąd)
Punkty uchwytu leków stosowanych w hormonoterapii raka gruczołu krokowego.
Autor: Nephron, Licencja: CC BY-SA 3.0
Micrograph showing prostate carcinoma with extraprostatic extension (EPE). Core biopsy. H&E stain.
In the context of a biopsy, EPE is defined as tumour adjacent to fat. The tumour seen is Gleason pattern 4.
Related images

EPE - intermed. mag.
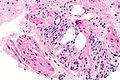
EPE - high mag.


Autor:
- Cancer_Research_UK_uploader
- derivative work: M1llx (zgłoś błąd)
Zabieg usunięcia prostaty.
Autor:
- Jonathan.Marcus, based on an original drawing by Dr. Marianne D Sadar (Meehan KL, Sadar MD. Front Biosci. 2003 May 1;8:d780-800).
- derivative work: M1llx (zgłoś błąd)
Funkcja receptora androgenowego. Testosteron (T) wchodzi do komórki i, jeśli występuje 5-alfa-reduktaza, ulega konwersji do dihydrotesteronu (DHT). Po związaniu steroidem, receptor androgenowy (AR) ulega zmianie konformacyjnej i uwalnia białka szoku cieplnego (hsp). Fosforylacja (F) występuje przed i / lub po związaniu steroidów. AR przemieszcza się do jądra, w którym dochodzi do dimeryzacji, wiązania DNA i rekrutacji koaktywatorów. Docelowe geny są transkrybowane (mRNA) i tłumaczone na białka (translacja).
Autor: Cancer Research UK uploader, Licencja: CC BY-SA 4.0
Biopsja gruczołu krokowego.
Skala Gleasona.
Autor: Nephron, Licencja: CC BY-SA 3.0
Micrograph showing Gleason pattern 4 in prostatic carcinoma. H&E stain
Related images
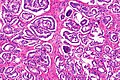
PCGP - intermed. mag.

PCGP - high mag.


Autor: Nephron, Licencja: CC BY-SA 3.0
High magnification micrograph of prostate adenocarcinoma. Needle core biopsy. HPS stain.
See also
- Image:Prostate adenocarcinoma 2 intermed mag hps.jpg - same case, intermediate magnification.
- Image:Prostate adenocarcinoma intermed mag hps.jpg - same case, different core.
- Image:Perineural invasion prostate high mag.jpg - same case, view demonstrating perineural invasion.
Autor: myself (Alex_brollo), Licencja: CC-BY-SA-3.0
Whole slide of half a prostate, scanned (Epson Perfection 1670 scanner)
Autor: Nephron, Licencja: CC BY-SA 3.0
Micrograph showing prostatic acinar adenocarcinoma (the most common form of prostate cancer) Gleason pattern 4 (left of image) and Gleason pattern 5 (right of image). H&E stain. Prostate currettings.
See also
Autor: Nephron, Licencja: CC BY-SA 3.0
Micrograph showing prostatic carcinoma with intraglandular blue mucin, a feature suggestive of malignancy. H&E stain.
The fields shown have a small amount of Gleason pattern 4 on the background of Gleason pattern 3.
Related images

PC with blue mucin - low mag.
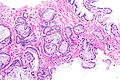
PC with blue mucin - intermed. mag.

PC with blue mucin - intermed. mag.

PC with blue mucin - intermed. mag.

PC with blue mucin - high mag.

PC with blue mucin - very high mag.






Autor: Nephron, Licencja: CC BY-SA 3.0
Micrograph showing ductal adenocarcinoma of the prostate gland, also prostatic ductal adenocarcinoma. H&E stain.
Related images

DAP - low mag.

DAP - intermed. mag.

DAP - high mag.

DAP - very high mag.




Autor:
- BruceBlaus
- derivative work: M1llx (zgłoś błąd)
Usuwanie prostaty.
Autor: RadsWiki, Licencja: CC BY-SA 3.0
Osseous mets from prostate cancer; bone scan
Autor: Nephron, Licencja: CC BY-SA 3.0
Micrograph of prostatic adenocarcinoma, conventional (acinar) type, the most common form of prostate cancer. Prostate biopsy. H&E stain.
Autor: James Heilman, MD, Licencja: CC BY-SA 4.0
Brachytherapy beads used to treat prostate cancer. Arrow marks beads.
Autor:
- Cancer Research UK uploader
- derivative work: M1llx (zgłoś błąd)
Brachyterapia o wysokiej mocy dawki.
Badanie per rectum.
Autor: Librepath, Licencja: CC BY-SA 3.0
Micrograph showing a biopsy of the rectum with prostatic carcinoma. H&E stain/PSAP/PSA/CK20.
Related images

Pca in rectum - very low mag.

Pca in rectum - low mag.

Pca in rectum - intermed. mag.

Pca in rectum - high mag.

Pca in rectum - very high mag.

Pca in rectum - intermed. mag.

Pca in rectum - high mag.








Pca in rectum - low mag.

Pca in rectum - intermed. mag.

Pca in rectum - high mag.

Pca in rectum - intermed. mag.

Pca in rectum - high mag.

Pca in rectum - very high mag.

Pca in rectum - low mag.

Pca in rectum - intermed. mag.








Autor:
- Cancer_Research_UK_uploader
- derivative work: M1llx (zgłoś błąd)
Brachyterapia o niskiej mocy dawki
Autor: James Heilman, MD, Licencja: CC BY-SA 4.0
Sclerosis of the bones of the thoracic spine due to prostate cancer metastases (CT image)
Autor: Yale Rosen from USA, Licencja: CC BY-SA 2.0
This is an example of lymphangitic carcinomatosis with metastatic tumor seen here primarily in subpleural lymphatics.
Photo of radioactive seeds. These implants are a form of radiation therapy for prostate cancer. Brachytherapy or internal radiation therapy are also terms used to describe this procedure
Visit the Nuclear Regulatory Commission's website at www.nrc.gov/.
To comment on this photo go to public-blog.nrc-gateway.gov/2012/04/01/nrc-moves-its-publ....
Photo Usage Guidelines: www.flickr.com/people/nrcgov/
Privacy Policy: www.nrc.gov/site-help/privacy.html.Autor: Narenfox, Licencja: CC BY-SA 4.0
Linear Accelerator - Elekta Compact model at Narayana Multispeciality Hospital, Mysore. Equipment is used for Radiotherapy
U.S. Army and Ghanaian medical professionals perform a radical prostatectomy during Medical Readiness Training Exercise 17-2 at the 37th Military Hospital in Accra, Ghana, Feb. 8, 2017. MEDRETE 17-2 includes participants from the Ghanaian government, U.S. Army Africa, Brooke Army Medical Center in San Antonio, Texas, and the North Dakota National Guard. It is the second in a series of medical readiness training exercises that USARAF is scheduled to facilitate in various countries in Africa. The mutually beneficial exercise offers opportunities for the partnered militaries to cooperate on medical specific tasks, share best practices and improve medical treatment processes. (U.S. Army Africa photo by Staff Sgt. Shejal Pulivarti)
- Unit: U.S. Army Africa
- DVIDS Tags: medical; U.S. Army Africa; MEDRETE; doctors; exercise; partnership; Ghana; training; North Dakota Army National Guard; Accra; Medical Readiness Training Exercise; Brooke Army Medical Center; USARAF; #USARAF_MEDRETE17; Medical Readiness Training Exercise 17-2
Autor: Nephron, Licencja: CC BY-SA 3.0
Micrograph of urothelial carcinoma (also urothelial cell carcinoma) within the prostate. H&E stain.
Related images

UCC in prostate - low mag.

UCC in prostate - intermed. mag.

UCC in prostate - high mag.

UCC in prostate - high mag.

UCC in prostate - intermed. mag.

UCC in prostate - high mag.
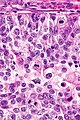
UCC in prostate - very high mag.

UCC in prostate - intermed. mag.

UCC in prostate - high mag.

UCC in prostate - very high mag.










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Osteoplastic bone metastasis of a histologically confirmed prostate cancer. Choline uptake in PET correlates with hyerdense structure. Axial display of CT, PET und PET/CT. SUV = 3,4