Rak jelita grubego
| ||
| carcinoma intestini crassi | ||
 Rak jelita grubego (preparat pooperacyjny) | ||
| ICD-10 | C18 Nowotwór złośliwy jelita grubego | |
| C18.0 | jelito ślepe | |
| C18.1 | wyrostek robaczkowy | |
| C18.2 | okrężnica wstępująca | |
| C18.3 | zgięcie wątrobowe | |
| C18.4 | okrężnica poprzeczna | |
| C18.5 | zgięcie śledzionowe | |
| C18.6 | okrężnica zstępująca | |
| C18.7 | esica | |
| C18.8 | zmiana przekraczająca granice jednego umiejscowienia w obrębie jelita grubego | |
| C18.9 | okrężnica, umiejscowienie nieokreślone | |
| ICD-10 | C19 Nowotwór złośliwy zgięcia esiczo-odbytniczego | |
| ICD-10 | C20 Nowotwór złośliwy odbytnicy | |
Rak jelita grubego (łac. carcinoma intestini crassi) – pierwotny nowotwór złośliwy jelita grubego wywodzący się z nabłonka błony śluzowej jelita grubego. Do raka jelita grubego zaliczany jest rak okrężnicy, rak zgięcia esiczo-odbytniczego i rak odbytnicy. Klinicznie bywa dzielony na raka okrężnicy i odbytnicy[1]. Rak kanału i brzegu odbytu jest osobną jednostką kliniczną[2].
Pod względem zapadalności na świecie stanowi drugi co do częstości nowotwór złośliwy u kobiet i trzeci u mężczyzn[3][4]. W 2012 roku rozpoznano 1 340 000 przypadków raka jelita grubego i 690 000 zgonów z jego powodu[5]. Histopatologicznie zdecydowaną większość nowotworów jelita grubego u ludzi stanowi gruczolakorak. Objawy choroby zależą od lokalizacji guza oraz jej zaawansowania. Początkowo nowotwór pozostaje bezobjawowy lub skąpoobjawowy. Typowym objawem raka jelita grubego jest krwawienie, które przy dalszej lokalizacji guza jest obserwowane jako jawna obecność krwi zmieszanej ze stolcem lub krwi na stolcu, a w przypadku bliższej lokalizacji guza krew ulega degradacji i świeża krew nie jest widoczna, a krwawienie objawia się obecnością ciemno zabarwionych stolców i niedokrwistości z niedoboru żelaza. Anemia manifestuje się przede wszystkim osłabieniem, ciągłym zmęczeniem oraz bladością skóry i błon śluzowych. Ból brzucha występujący przy raku jelita grubego jest niecharakterystyczny, jego lokalizacja i typ zależą od lokalizacji guza. Mogą występować naprzemienne biegunki i zaparcia, a także spadek masy ciała. W zaawansowanej chorobie w badaniu fizykalnym może być wyczuwalny guz, powiększenie wątroby lub węzłów chłonnych[6].
Duże znaczenie w powstawaniu choroby mają modyfikowalne czynniki ryzyka, z których największe znaczenie ma nieprawidłowa dieta ze zbyt dużą ilością czerwonego mięsa, tłuszczów nasyconych i zbyt małą ilością warzyw, owoców i błonnika. Istotne znaczenie mają palenie tytoniu, otyłość, cukrzyca i nieswoiste zapalenia jelit. W 20–30% przypadków raka zachorowania mają tło dziedziczne, które zwykle pozostaje niezidentyfikowane[7].
Podstawowym badaniem pozwalającym rozpoznać raka jelita grubego jest kolonoskopia, która umożliwia uwidocznienie guza i pobranie próbek do badania histopatologicznego. Pomocniczo stosuje się wirtualną kolonoskopię TK lub MRI, endoskopię kapsułkową oraz radiologiczne badanie dwukontrastowe. W ocenie zaawansowania stosuje się tomografię komputerową, rezonans magnetyczny, ultrasonografię endoskopową oraz pozytonową tomografię emisyjną[8]. Zaawansowanie choroby klasyfikuje się w klasyfikacji TNM[9].
Leczenie raka jelita grubego warunkuje zaawansowanie choroby oraz stan chorego. Wykonuje się zabieg operacyjny, który ma na celu usunięcie guza wraz z marginesem zdrowych tkanek. Rodzaj zabiegu zależy od lokalizacji guza i zwykle polega na resekcji części jelita z usunięciem regionalnych węzłów chłonnych w jednym bloku tkankowym z odtworzeniem ciągłości przewodu pokarmowego[10]. W przypadku guza położonego w odbytnicy, dąży się również do odtworzenia ciągłości przewodu pokarmowego z zachowaniem zwieraczy odbytnicy, jednakże w przypadku zbyt niskiego położenia guza, konieczna jest amputacja odbytnicy i wytworzenie stałej kolostomii[11]. Leczenie operacyjne w zaawansowanych przypadkach raka odbytnicy jest poprzedzone leczeniem neoadiuwantowym[12], a w zaawansowanych przypadkach raka okrężnicy jest uzupełnione leczeniem adiuwantowym[10]. Leczenie choroby z przerzutami jest oparte na chemioterapii, której charakter zależy od założonych celów terapeutycznych i stanu chorego[13]. U niewielkiej części chorych możliwe jest chirurgiczne usunięcie pojedynczych przerzutów do wątroby lub płuc[14]. W chemioterapii wielolekowej stosuje się programy składające się z fluoropirymidyn (5-fluorouracylu, kapecytabiny) w połączeniu z oksaliplatyną lub irynotekanem[15].
Objawy

jelito ślepe (kątnica)
okrężnica wstępująca (wstępnica)
okrężnica poprzeczna (poprzecznica)
okrężnica zstępująca (zstępnica)
okrężnica esowata (esica)
odbytnica
Objawy raka jelita grubego zależą od lokalizacji guza i stopnia zaawansowania choroby[16]. Objawy wynikają z miejscowego wzrostu guza w obrębie światła jelita, nacieku sąsiednich struktur oraz obecności przerzutów odległych[17]. Przebieg choroby może być przez długi czas bezobjawowy[6][18].
Przewlekłe krwawienie z przewodu pokarmowego jest jednym z najczęstszych objawów raka jelita grubego i jest stwierdzane u większości chorych. Jawna obecność krwi występuje przy guzach położonych w dalszej części jelita grubego[17]. Rak odbytnicy może powodować obecność świeżej krwi na stolcu, z kolei rak zstępnicy i esicy może być przyczyną obecności krwi zmieszanej ze stolcem lub na stolcu (hematochezja)[19]. Krew w stolcu stwierdza się u około 80% chorych z rakiem odbytnicy, 45% chorych z rakiem w lewej połowie okrężnicy i 20% prawej połowy okrężnicy[20]. W przypadku guzów położonych w prawej części jelita grubego zwykle nie stwierdza się jawnej obecności krwi w stolcu, co jest związane z degradacją i wymieszaniem krwi ze stolcem podczas pasażu przez dalszą część jelita grubego. Obecność utajonego krwawienia skutkuje niedokrwistością z niedoboru żelaza objawiającą się osłabieniem, łatwą męczliwością, dusznością, bladością skóry i błon śluzowych oraz przyspieszeniem akcji serca, a także obniżeniem stężenia hemoglobiny stwierdzanym w morfologii krwi[17]. Utajone krwawienie jest stwierdzane aż u 80% chorych na raka jelita grubego, a obniżenie stężenia hemoglobiny u 60% chorych[21]. Domieszka krwi z bliższej części okrężnicy, poddanej rozkładowi przez bakterie, skutkuje obecnością smolistego stolca (stolec o ciemnym zabarwieniu)[22].
Ból brzucha lub dyskomfort w brzuchu jest częstym objawem raka jelita grubego i występuje u około połowy chorych[21]. Ból brzucha towarzyszący rakowi jelita grubego może być kurczowy, kolkowy lub stały[18]. Lokalizacja bólu jest różna w zależności od lokalizacji choroby i może występować w okolicy podbrzusza, nadbrzusza, okolicy pępkowej oraz okolicy kroczowej, choć często ból jest trudny do zlokalizowania[23][18].
Zmiana rytmu wypróżnień z naprzemiennymi zaparciami i biegunkami z domieszką śluzu może być spowodowana rakiem[18][16], jednak jest to stosunkowo częsty objaw w populacji ogólnej[24]. Rakowi odbytnicy może towarzyszyć uczucie niepełnego oddawania stolca[24], a także zmniejszenie szerokości oddanego stolca nazywane stolcami ołówkowatymi[19].
Niezamierzony spadek masy ciała występuje u około połowy chorych na raka jelita grubego. Częściowo jest on spowodowany lub nasilany uciążliwymi objawami ze strony jamy brzusznej, które prowadzą do zmniejszenia apetytu. U chorych z zaawansowaną chorobą za utratę masy ciała odpowiada zespół kacheksja-anoreksja[25].
Wzrost guza w kierunku światła jelita grubego może spowodować zwężenie światła i wywołać przepuszczającą lub całkowitą niedrożność jelit. Niedrożność częściej pojawia się w esicy i odbytnicy. Częściowa niedrożność jelita grubego objawia się kolkowym bólem brzucha, zatrzymaniem stolca, wzdęciem i nudnościami. Ból kolkowy pojawia się z mniejszą częstością i mniejszą intensywnością niż w niedrożności jelita cienkiego. Brzuch podczas badania palpacyjnego jest nieco bolesny, w osłuchiwaniu jamy brzusznej mogą być słyszalne dźwięki perystaltyki, jednak w późniejszym etapie perystaltyka zanika[26][17]. W niedrożności całkowitej pojawiają się ból brzucha, zatrzymanie stolca i gazów, nudności oraz wymioty[17]. Masywny krwotok do przewodu pokarmowego lub perforacja zdarzają się stosunkowo rzadko[16].
W badaniu palpacyjnym brzucha w zaawansowanym stadium choroby bywa wykrywany guz. Częściej jednak stwierdza się powiększenie wątroby i wodobrzusze związane z obecnością przerzutów[6]. Badanie per rectum (przez odbyt) często pozwala palpacyjnie rozpoznać obecność guza, obecność krwi, rzadziej próg Blumera związany z obecnością masy nowotworowej w zachyłku odbytniczo-pochwowym u kobiet lub zachyłku odbytniczo-pęcherzowym u mężczyzn[6][25]. Bywają obecne powiększone węzły chłonne okolicy okołopępkowej (guz siostry Mary Joseph) i powiększenie węzłów chłonnych nadobojczykowych (węzeł Virchowa)[25].
Czynniki ryzyka
Do czynników ryzyka zachorowania na raka jelita grubego należą[27]:
- nieprawidłowa dieta zbyt bogata w czerwone mięso, szczególnie smażone i grillowane, tłuszcze nasycone i zbyt uboga w świeże warzywa i owoce,
- nadwaga i otyłość,
- niska aktywność fizyczna,
- palenie tytoniu,
- cukrzyca,
- czynniki genetyczne:
- występowanie raka jelita grubego w rodzinie,
- zespół Lyncha,
- rodzinny rak jelita grubego typu X,
- zespół rodzinnej polipowatości gruczolakowatej,
- zespół Peutza-Jeghersa,
- polipowatość związana z mutacją MYH,
- nieswoiste zapalenia jelit,
- nadmierne spożywanie alkoholu,
- akromegalia,
- radioterapia w obrębie miednicy.
Ochronną rolę wykazują warzywa i owoce oraz prawdopodobnie spożycie błonnika i kwasów tłuszczowych omega-3[27].
Dieta

Nieprawidłowa dieta ma kluczowy wpływ na rozwój raka jelita grubego[28][29]. Może ona odpowiadać za 70–90% wszystkich jego przypadków. Nadmierne spożywanie czerwonego mięsa i tłuszczów zwierzęcych sprzyja rozwinięciu raka jelita grubego. Prawdopodobnie spożycie warzyw i owoców wykazuje działanie chroniące przed rakiem jelita grubego. Ochronna rola kwasów tłuszczowych omega-3 i błonnika jest niejasna[30].
- Nadmierne spożycie czerwonego mięsa
Nadmierne spożycie czerwonego mięsa, szczególnie poddanego smażeniu lub grillowaniu jest czynnikiem ryzyka rozwoju raka jelita grubego[30][31]. Metaanaliza wykazała, że nadmierne spożycie czerwonego mięsa zwiększa ryzyko rozwoju raka jelita grubego o 33%[32]. Zwiększenie dziennej konsumpcji czerwonego mięsa o 100 g zwiększa ryzyko rozwoju raka o 12–17%[33]. Wykazano, że ryzyko rozwoju raka jest większe w przypadku smażenia lub grillowania czerwonego mięsa[34][35][30].
Jednak kilka badań podważa związek spożycia czerwonego mięsa ze zwiększonym ryzykiem zachorowania ze względu na trudności z oddzieleniem wpływu innych czynników ryzyka związanych ze stylem życia oraz samym sposobem obróbki cieplnej mięsa (w tym smażeniem i grillowaniem mięsa)[36][37][30].
- Nadmierne spożycie tłuszczów pochodzenia zwierzęcego
Prawdopodobnie nadmierne spożycie tłuszczów nasyconych, które są obecne głównie w produktach pochodzenia zwierzęcego, jest czynnikiem zwiększonego ryzyka zachorowania na raka jelita grubego[30]. Kilka badań wskazuje na wzrost ryzyka zachorowania spowodowany wysokim spożyciem tłuszczów pochodzenia zwierzęcego[38][39]. Na ryzyko związane z tłuszczami nasyconymi wskazały dwa duże badania. Badanie przeprowadzone na grupie 80 000 kobiet potwierdziło ryzyko związane z nadmiernym spożyciem tłuszczów pochodzenia zwierzęcego i wspiera zalecenie zastąpienia czerwonego mięsa drobiem i rybami[39]. Badanie przeprowadzone na grupie 130 000 osób wskazało na 20% obniżenie ryzyka zachorowania na raka u osób, które zmniejszyły spożycie tłuszczów pochodzenia zwierzęcego[40]. Z drugiej strony jedna metaanaliza kilku badań nie wykazała związku pomiędzy spożyciem tłuszczów pochodzenia zwierzęcego a ryzykiem rozwoju raka jelita grubego[41].
- Kwasy tłuszczowe omega-3
Prawdopodobnie kwasy tłuszczowe omega-3 mogą zapobiegać zachorowaniu na raka jelita grubego, choć liczba dowodów naukowych jest ograniczona[30]. Kilka badań wskazało na zmniejszone ryzyko rozwoju raka u osób o zwiększonym spożyciu kwasów tłuszczowych omega-3[42][43][44][45]. W chińskiej metaanalizie spożycie tłustych ryb, które są bogatym źródłem kwasów tłuszczowych omega-3, zmniejsza ryzyko pojawienia się raka jelita grubego o 12%[46].
Z drugiej strony są badania zaprzeczające lub niewspierające korelacji spożycia kwasów omega-3 i redukcji ryzyka zachorowania na raka jelita grubego[47][48]. Prawdopodobnie sztuczna suplementacja kwasów tłuszczowych omega-3 nie zmniejsza ryzyka rozwinięcia raka jelita grubego[49].
- Warzywa i owoce

Prawdopodobnie ryzyko zachorowania na raka jelita grubego zmniejsza spożycie warzyw i owoców[31][50]. Ochronne działanie może być rezultatem obecności antyoksydantów, błonnika, witamin, w tym kwasu foliowego, mikroelementów i flawonów[31]. Dieta bogata w warzywa, owoce i otręby pszenne zmniejsza ryzyko powstania gruczolaków, które są prekursorem większości przypadków raka jelita grubego[51][52][53][54][55][30].
- Błonnik
Rola błonnika w zapobieganiu zachorowaniu na raka jelita grubego jest niejasna[30]. Starsze badania wskazywały na ochronne działanie błonnika[56][57]. Jednak kilka późniejszych badań nie potwierdzało tego efektu lub wykazywało niewielki efekt ochronny[30]. Metaanaliza z 2005 roku nie udowodniła ochronnego wpływu błonnika[58], ale późniejsza metaanaliza z 2011 roku wskazała, że spożycie błonnika redukuje ryzyko raka jelita grubego i zwiększenie spożycia błonnika o 10 g dziennie zmniejsza ryzyko raka o 10%[59].
- Witamina D3 i wapń
Dane dotyczące ochronnego wpływu witaminy D3 i wapnia są niespójne, choć niektóre badania sugerują ich korzystne działanie[30]. Ochronne działanie witaminy D3 może wynikać ze stymulacji różnicowania się komórek i hamowania ich podziałów[30]. Przesłanką do ochronnej roli witaminy D3 jest wyższa śmiertelność na raka jelita grubego w regionach o wyższej szerokości geograficznej, gdzie z powodu mniejszego nasłonecznienia synteza witaminy jest słabsza[60] oraz niższa częstość występowania raka w populacjach o wysokim spożyciu świeżych ryb, skorupiaków i witaminy D[61][30].
Wyniki badań roli witaminy D3 są niespójne. Jedno badanie wskazuje na odwrotny stosunek stężenia witaminy D3 i ryzyka raka oraz redukcję ryzyka raka o 50% przy suplementacji witaminy w dawce 1000 IU dziennie[62], z kolei inne nie wykazują ochronnego działania witaminy D3[63][64].
Podobnie niejasna jest rola wapnia. Kilka badań wskazuje na działanie ochronne przy spożyciu 700–800 mg dziennie[65][66], a kilka nie stwierdza tego efektu[67][68]. Jednak metaanaliza 10 badań wskazuje na 22% redukcję ryzyka zachorowania na raka jelita grubego[69].
Otyłość

Nadwaga i otyłość są niezależnymi czynnikami ryzyka wystąpienia raka jelita grubego. W dużym badaniu wskaźnik BMI 30 jest związany z 1,19-krotnie zwiększonym ryzykiem zachorowania na raka. Ryzyko rozwoju raka jest proporcjonalne do stopnia otyłości. Wzrost wskaźnika BMI o 2 zwiększa ryzyko o 7%, a wzrost obwodu talii o 2 cm zwiększa ryzyko o 4%[70]. Nadwaga i otyłość jest związana z 11% przypadków raka jelita grubego. Ryzyko raka może być zwiększone o 30–70%[71]. Otyłość jest czynnikiem wpływającym na zwiększenie ryzyka rozwoju raka okrężnicy u mężczyzn i kobiet, przy czym ryzyko jest znacząco większe u mężczyzn. Ryzyko rozwoju raka odbytnicy jest większe u mężczyzn, ale u kobiet nie zaobserwowano korelacji otyłości z ryzykiem wystąpienia raka odbytnicy[72][73]. Wykazano, że otyłość brzuszna zwiększa ryzyko pojawienia się gruczolaka w porównaniu do osób bez otyłości[74][75].
Cukrzyca
Cukrzyca jest czynnikiem zwiększającym ryzyko zachorowania na raka jelita grubego[76][77][78][79]. Ryzyko dotyczy obu płci i lokalizacji w okrężnicy oraz odbytnicy[80]. Zwiększone ryzyko prawdopodobnie jest rezultatem stymulacji wzrostu komórek przez insulinę i IGF oraz ułatwienia wykorzystania glukozy przez komórki nowotworowe[79][31][81]. Ponadto wykazano, że chorzy z cukrzycą mają gorsze rokowanie i wyższe ryzyko zgonu z powodu raka jelita grubego niż chorzy bez cukrzycy[82].
Niska aktywność fizyczna
Niska aktywność fizyczna wiąże się ze zwiększonym ryzykiem wystąpienia raka jelita grubego[83][31]. U 25–33% chorych na raka jelita grubego stwierdza się powiązane modyfikowalne czynniki ryzyka: otyłość i brak aktywności fizycznej[28].
Istnieją dowody sugerujące, że wzrost aktywności fizycznej zmniejsza ryzyko zachorowania[84][85][28]. Zaobserwowano relację dawka–odpowiedź, a zatem większa częstość i intensywność aktywności fizycznej przekłada się na zmniejszenie ryzyka zachorowania[86][28]. Aktywność fizyczna redukuje ryzyko prawdopodobnie poprzez przyspieszenie tempa metabolizmu, obniżenie masy ciała i insulinooporności oraz przyspieszenie perystaltyki jelit[28].
Palenie tytoniu
Palenie tytoniu zwiększa ryzyko zachorowania na raka jelita grubego[87][28][31]. Zwiększa ono 1,25-krotnie względne ryzyko zachorowania, a także zwiększa ryzyko zgonu z powodu raka jelita grubego[88]. Ryzyko jest proporcjonalne do dziennej ilości wypalanych papierosów, lat trwania nałogu, paczkolat, a także w mniejszym stopniu niższego wieku rozpoczęcia palenia[89]. Palenie wykazuje większy związek z rakiem okrężnicy niż odbytnicy[89][90]. Zaobserwowano związek palenia z większym ryzykiem powstawania polipów w jelicie grubym[91]. Po zaprzestaniu palenia spadek ryzyka zachorowania do ryzyka populacyjnego obserwuje się dopiero po 30 latach od porzucenia nałogu[92].
Nieswoiste zapalenia jelit

Do nieswoistych zapaleń jelita związanych ze zwiększonym ryzykiem zachorowania na raka jelita grubego należą wrzodziejące zapalenie jelita grubego i choroba Leśniowskiego-Crohna. Ryzyko jest uzależnione od typu zapalenia jelita, długości trwania choroby i jej rozległości[93]. Metaanaliza długoterminowych badań z 2001 roku wykazała, że ryzyko rozwoju raka u osób chorujących na wrzodziejące zapalenie jelita grubego wynosi po 10 latach choroby 1,6%, po 20 latach już 8,3%, a po 30 latach choroby 18,4%[94]. Późniejsze wyniki europejskich badań wskazały na niższe ryzyko rozwoju raka[95][93]. Z kolei metaanaliza badań oceniających ryzyko rozwoju raka u chorych z chorobą Leśniowskiego-Crohna wskazała na porównywalne, choć niższe ryzyko w stosunku do wrzodziejącego zapalenia jelita grubego[93][93]. Ryzyko po 10 latach w chorobie Leśniowskiego-Crohna wynosi 2,9%, po 20 latach 5,6% i 8,3% po 30 latach choroby[96][93].
Ryzyko zachorowania na raka jest większe w przypadku rozległego zajęcia jelita grubego u chorych z wrzodziejącym zapaleniem jelita grubego. Niewielkie lub wręcz bez zwiększonego ryzyka dotyczy zajęcia odbytnicy lub esicy. Pośrednie ryzyko dotyczy zajęcia lewej połowy okrężnicy, a największe zajęcia całego jelita grubego (pancolitis)[94][97][95][98][93]. Pierwotne stwardniające zapalenie dróg żółciowych (PSC) jest pozajelitową manifestacją nieswoistych zapaleń jelit, częściej występujące przy wrzodziejącym zapaleniu jelita grubego. Pierwotne stwardniające zapalenie dróg żółciowych znacznie zwiększa ryzyko wystąpienia raka jelita grubego. Wykazano, że 21% chorych z pierwotnym stwardniającym zapaleniem dróg żółciowych i nieswoistym zapaleniem jelit rozwija raka jelita grubego[99]. Występowanie sporadycznego raka jelita grubego w rodzinie chorego na nieswoiste zapalenie jelit dodatkowo zwiększa ryzyko zachorowania na raka[100][101].
Alkohol
Nadmierne spożywanie alkoholu etylowego niesie ze sobą zwiększone ryzyko raka jelita grubego[102]. Ryzyko jest proporcjonalne do ilości spożywanego alkoholu[103]. Wykazano, że u osób spożywających ponad 45 g etanolu dziennie występuje 1,4-krotnie zwiększone ryzyko zachorowania. Zwiększone ryzyko, choć mniejsze, jest obserwowane również przy spożyciu 30–40 g alkoholu[104][31].
Napromieniowywanie miednicy
Prawdopodobnie wcześniejsza radioterapia w obrębie miednicy wiąże się ze zwiększonym ryzykiem zachorowania[85][105][31].
Akromegalia
Chorzy na akromegalię są bardziej narażeni na pojawienie się gruczolaków jelita grubego[106] oraz raka jelita grubego[107][31].
Występowanie rodzinne i czynniki genetyczne
W 20–30% przypadków raka jelita grubego obserwuje się jego rodzinne występowanie[108][109][110]. W 3–5% przypadków jest ono spowodowane definiowalnymi zespołami zwiększonej predyspozycji do nowotworów[108][109][111][110].
- Zespół Lyncha (dziedziczny rak jelita grubego niezwiązany z polipowatością, HNPCC)
Zespół Lyncha jest zespołem zwiększonej predyspozycji genetycznej do nowotworów złośliwych, jest dziedziczony autosomalnie dominująco i uwarunkowany mutacjami genów odpowiedzialnych za naprawę DNA, najczęściej genów MSH2 i MLH1. Zespół Lyncha stanowi około 2–3% wszystkich raków jelita grubego[112][113][111][114]. Wiąże się z 80% ryzykiem zachorowania na raka jelita grubego[115][110]. Rak wykazuje skłonność do pojawiania się w prawej (proksymalnej) części jelita grubego, cechowania się niższym stopniem zróżnicowania (wyższy stopień złośliwości histologicznej) oraz komponentem śluzowym[116]. Zwykle rak jelita grubego pojawia się między 40. a 50. rokiem życia, średni wiek zachorowania wynosi 44 lata[117][116]. U około jednej trzeciej chorych rozwijają się inne nowotwory niż rak jelita grubego[111]. U kobiet najczęściej jest to rak trzonu macicy, którego ryzyko wystąpienia w ciągu życia wynosi 40–60%[116]. Ryzyko zachorowania na raka żołądka w ciągu życia wynosi około 13%, a na raka jajnika około 12%[118]. W zespole Lyncha obserwuje się również zwiększone ryzyko rozwoju raka jelita cienkiego, raka nerki, raka moczowodu, a rzadziej raka podstawnokomórkowego skóry, raka dróg żółciowych i nowotworów centralnego układu nerwowego[119][120][118][121].
- Rodzinny rak jelita grubego typu X (Familial Colorectal Cancer Type X, FCCX)
Rodzinny rak jelita grubego typu X to zespół, w którym obserwuje się rodzinnie zwiększone ryzyko zachorowania na raka jelita grubego i spełnione są kryteria amsterdamskie, ale nie rozpoznaje się niedoborów lub defektu genów naprawy DNA[122][110][123]. Zgodnie z kryteriami amsterdamskimi stwierdza się obecność co najmniej trzech członków rodziny chorych na raka jelita grubego, z czego jeden krewny wykazuje pokrewieństwo I stopnia w stosunku do pozostałych chorych, rak pojawia się w co najmniej dwóch pokoleniach i co najmniej jedno zachorowanie pojawia się przed 50. rokiem życia. Obserwuje się dziedziczenie autosomalne dominujące, jednak podłoże genetyczne pozostaje niepoznane[124]. Kryteria amsterdamskie, ale bez rozpoznanej mutacji lub niedoborów genów naprawy DNA spełnia 40–50% rodzin[110][122].
Rak pojawia się w późniejszym wieku niż w zespole Lyncha i ryzyko pojawienia się raka u krewnych jest niższe niż zespole Lyncha[110]. W zespole nie obserwuje się zwiększonego ryzyka występowania nowotworów innych niż rak jelita grubego[122].
- Rodzinna polipowatość gruczolakowata (FAP)

Rodzinna polipowatość gruczolakowata jest zespołem predyspozycji do nowotworów dziedziczonym autosomalnie dominująco, uwarunkowanym mutacją w genie APC. Zespół charakteryzuje się bardzo licznymi gruczolakami na całej długości jelita grubego oraz cienkiego, a także w żołądku. Gruczolaki predysponują do rozwoju raka jelita grubego, którego ryzyko jest bliskie 100%[125]. Stanowi drugi pod względem częstości występowania zespół predyspozycji do raka jelita grubego, odpowiada za około 1% wszystkich przypadków raka w tej lokalizacji[108]. Polipy są obecne u połowy chorych w wieku 15 lat i u 95% chorych w wieku 35 lat[126]. Liczba polipów może się wahać od kilkuset do kilku tysięcy[108]. Zwykle rak rozwija się około 35. roku życia, choć rzadko bywa obecny przed 20. rokiem życia[126]. Zespołowi rodzinnej polipowatości gruczolakowatej towarzyszą również inne nowotwory złośliwe i łagodne. Należą do nich rak tarczycy, gruczolakorak jelita cienkiego, hepatoblastoma, rak trzustki, rak żołądka, rak nadnerczy i gruczolaki nadnerczy, guzy mózgu (głównie rdzeniak)[126].
- Zespół Peutza-Jeghersa
Zespół Peutza-Jeghersa jest zespołem zwiększonej predyspozycji genetycznej do nowotworów złośliwych dziedziczonym autosomalnie dominująco i uwarunkowanym mutacją genu supresorowego LKB1 (STK11). W zespole Peutza-Jeghersa występują hamartomiczne polipy w przewodzie pokarmowym, melanoza skóry i błon śluzowych głównie okolicy warg ust, jamy ustnej, narządów płciowych, kończyn oraz odbytu. Zwiększona predyspozycja w różnym stopniu dotyczy wzrostu ryzyka zachorowania na raka jelita cienkiego, raka żołądka, raka jelita grubego, raka trzustki, raka przełyku, raka piersi, raka szyjki macicy, raka trzonu macicy, raka jajnika, raka płuca i raka jądra[127][128]. Ryzyko pojawienia się złośliwego nowotworu przewodu pokarmowego w ciągu życia chorego wynosi 70%, a łącznie z pozostałymi nowotworami 90%[129][130]. Ryzyko raka jelita grubego w wieku 40 lat wynosi 3%, w wieku 50 lat 5%, w wieku 60 lat 15% i 39% w wieku 70 lat[129]. Zespół może odpowiadać za 0,01% wszystkich przypadków raka jelita grubego[131].
- Polipowatość młodzieńcza
Polipowatość młodzieńcza jest rzadkim zespołem predyspozycji do nowotworów dziedziczonym autosomalnie dominująco o niepełnej penetracji genu, najczęściej uwarunkowanym mutacjami w genach SMAD4 (MADH4) lub BMPR1A. W zespole występują hamartomiczne polipy najczęściej dotyczące jelita grubego (98% przypadków), ale również żołądka w 14% przypadków, jelita czczego i krętego w 7% i dwunastnicy w 2% przypadków[132][130]. Ryzyko rozwinięcia raka jelita grubego w wieku 35 lat wynosi 17–22%, a ryzyko rozwoju raka żołądka w ciągu życia wynosi 10–21%[131][130].
- Polipowatość związana z mutacją MYH (MAP)
Polipowatość związana z mutacją MYH jest rzadkim zespołem predyspozycji do nowotworów dziedziczonym autosomalnie recesywnie, związanym z mutacją genu MYH naprawiającego DNA. Klinicznie naśladuje późno pojawiający się zespół polipowatości rodzinnej[133]. Zwykle stwierdza się około 100 polipów. Zespół niesie ze sobą 70–80% ryzyko zachorowania na raka jelita grubego, który zwykle pojawia się około 50. roku życia. Prawdopodobnie nowotwory poza jelitem grubym nie są charakterystycznym elementem zespołu, jednak mogą one wystąpić[134].
Zapobieganie i badania przesiewowe





Profilaktyka raka jelita grubego jest oparta o walkę z modyfikowalnymi czynnikami ryzyka i program badań przesiewowych.
Profilaktyka pierwotna
Profilaktyka pierwotna obejmuje zwalczanie czynników ryzyka choroby. Profilaktyka pierwotna raka jelita grubego jest oparta na wpływie na modyfikowalne czynniki ryzyka. Kluczowa jest zmiana błędów dietetycznych polegających na zbyt wysokim spożyciu czerwonego mięsa i nasyconych kwasów tłuszczowych pochodzących głównie ze zwierzęcych produktów spożywczych oraz zbyt niskim spożyciu warzyw i owoców. Szczególnie smażone i grillowane czerwone mięso jest obarczone sprzyjaniem rozwojowi raka jelita grubego[30]. Zaleca się redukcję spożycia czerwonego mięsa oraz tłuszczów pochodzenia zwierzęcego i zamianę ich na drób, ryby i tłuszcze roślinne[135]. Warzywa i owoce wykazują działanie ochronne w raku jelita grubego i mogą w pewnym stopniu redukować ryzyko rozwoju raka[30].
Zaleca się zaprzestanie palenia tytoniu, które jest istotnym czynnikiem ryzyka rozwoju raka jelita grubego. Palenie nie tylko zwiększa ryzyko raka jelita grubego, ale również chorób układu krążenia, w tym zawału mięśnia sercowego i innych nowotworów złośliwych, w tym raka płuc, raka sutka, raka trzustki, raka pęcherza moczowego i nowotworów głowy i szyi. Szczególnie istotne jest zniechęcanie do palenia młodzieży i młodych dorosłych oraz zachęcanie do rzucenia palenia, ponieważ spadek ryzyka zachorowania obserwuje się dopiero po wielu latach od zaprzestania palenia tytoniu[92][135].
Ważna jest walka z otyłością, cukrzycą i zbyt niską aktywnością fizyczną. Otyłość typu centralnego, błędy dietetyczne, hiperinsulinemia związana z insulinoopornością oraz siedzący tryb życia sprzyjają rozwojowi raka jelita grubego. Zaleca się utrzymanie prawidłowej wagi i regularną aktywność fizyczną[136].
Profilaktyka wtórna
Profilaktyka wtórna obejmuje wczesne rozpoznawanie zmian przednowotworowych na podstawie badań przesiewowych. Rak jelita grubego rozwija się dość powoli na bazie łagodnych polipów, co daje możliwość wczesnego wykrycia i likwidacji zmian przednowotworowych lub wczesnego wykrycia i skutecznego leczenia niezaawansowanego raka[137]. Podstawowym celem badania przesiewowego jest redukcja śmiertelności z powodu choroby nowotworowej. Badania przesiewowe w raku jelita grubego obejmują badanie na obecność krwi utajonej w kale lub kolonoskopię[138].
- Strategie badań przesiewowych
W różnych krajach przyjęto różne strategie badań przesiewowych raka jelita grubego. Polegają one na badaniu na obecność krwi utajonej w kale wykonywanym co 1–2 lata lub kolonoskopii wykonywanej co 10 lat. Badanie przesiewowe jest zalecane u chorych powyżej 50. roku życia[138]. W większości krajów Europejskich badaniem przesiewowym jest badanie na obecność krwi utajonej w kale metodą immunochromatograficzną. W Stanach Zjednoczonych, Niemczech, Polsce i Włoszech w ramach badań przesiewowych wykonuje się kolonoskopię[137]. W Polsce program badań przesiewowych obejmuje osoby w wieku 50–65, bez krwawienia z przewodu pokarmowego, biegunki, zaparć, anemii lub utraty masy bez znanej przyczyny w ciągu ostatnich miesięcy, które mogą być wskazaniami do diagnostycznej, a nie przesiewowej kolonoskopii. W przypadku występowania raka u członka rodziny w pierwszym stopniu pokrewieństwa badanie przesiewowe wykonuje się również u osób w wieku 40–49 lat[137].
- Kolonoskopia
Kolonoskopia jest badaniem endoskopowym umożliwiającym zobrazowanie całej śluzówki jelita grubego i wykonanie niektórych zabiegów, w tym pobranie biopsji i usunięcie polipa. Przesiewowa kolonoskopia powinna obejmować inspekcję całego jelita grubego aż do kątnicy. Kolonoskopia powinna być wykonana z wykonaniem dożylnej sedacji, która redukuje nieprzyjemne odczucia związane z zabiegiem. Konieczne jest dobre przygotowanie do badania poprzez podanie odpowiednich środków przeczyszczających i właściwe nawodnienie[138]. Kolonoskopia pozwala oprócz wczesnego wykrycia raka na zmniejszenie częstości występowania tej choroby poprzez rozpoznawanie i usuwanie polipów, na bazie których może rozwinąć się rak inwazyjny[139][140][141][138]. Wykazano, że usunięcie polipów redukuje częstość występowania raka o 75–90%[138].
- Badanie na obecność krwi utajonej w kale
Coroczne badanie na obecność krwi utajonej w kale jest jedną z możliwych strategii badań przesiewowych. Badanie pozwala na wykrycie obecności niewidocznej w kale krwi, która może być związana z obecnością krwawiącego guza w przewodzie pokarmowym, ale również innych patologii prowadzących do krwawienia. Stwierdzenie obecności krwi w kale skłania do pogłębienia diagnostyki, przede wszystkim do wykonania kolonoskopii, która może potwierdzić lub wykluczyć chorobę nowotworową. Podstawowym ograniczeniem badania jest konieczność występowania krwawienia z guza[142]. Metaanaliza kilku badań wykazała, że badanie na obecność krwi utajonej w kale co roku lub co dwa lata o 16% zmniejsza śmiertelność na raka jelita grubego[143].
Badanie na obecność krwi w kale metodą gwajakolową cechuje się jednak stosunkowo niską czułością diagnostyczną dla wykrywania raka jelita grubego sięgającą 25–40% i również niską czułością w wykrywaniu zaawansowanych gruczolaków (16–30%)[144][138]. Konieczne jest pobranie trzech próbek i zastosowanie się do zaleceń niespożywania produktów spożywczych zawierających hemoglobinę[144]. Badanie na obecność krwi w kale metodą gwajakolową daje fałszywe wyniki w przypadku krwawienia do przewodu pokarmowego z innych powodów niż rak[144].
Metoda immunochromatograficzna jest oparta na wykrywaniu ludzkiej hemoglobiny. Zatem metoda nie wymaga zachowania odpowiedniej diety przed badaniem i jest niewrażliwa na krwawienie z górnej części przewodu pokarmowego, ponieważ hemoglobina ulega tam rozkładowi. Metoda jest też bardziej czuła niż metoda gwajakolowa[142]. Czułość metody immunochromatograficznej w wykrywaniu raka wynosi 60–90%, a dla wykrywania gruczolaków wynosi 30–70%[144].
Nadzór nad szczególnymi grupami ryzyka
- Zespół Lyncha
Zaleca się przeprowadzanie regularnej kolonoskopii co 1–2 lata od 20–25. roku życia. Badanie przesiewowe w kierunku raka endometrimu i raka jajnika rozpoczyna się 30–35. roku życia i polega na badaniu ginekologicznym, wykonaniu USG i badaniu CA 125. W profilaktyce raka żołądka zaleca się eradykację Helicobacter pylori i gastroskopię co 1–3 lata. Prawdopodobnie ryzyko rozwoju raka zmniejsza przyjmowanie kwasu acetylosalicylowego, choć dawka i długość leczenia nie zostały ustalone. Profilaktyczna kolektomia nie jest zalecana, ale w przypadku pojawienia się raka jelita grubego należy rozważyć rozszerzoną kolektomię ze względu na ryzyko wystąpienia nowotworów synchronicznych i metachronicznych[122].
- Rodzinny rak jelita grubego typu X
Zaleca się przeprowadzania regularnej kolonoskopii co 3–5 lat zaczynając je 5–10 lat wcześniej niż najwcześniejszy przypadek raka w rodzinie[122].
- Zespół rodzinnej polipowatości gruczolakowatej
W rodzinach z rozpoznaną mutacją genu APC zaleca się kolonoskopię co 2 lata od 18–20. roku życia. W rodzinach bez rozpoznanej mutacji genu APC wykonuje się sigmoidoskopię lub kolonoskopię co 2 lata aż do 40. roku życia, następnie co 3–5 lat do 50. roku życia i wówczas nadzór może być przerwany jeśli nie stwierdzono polipów. W przypadku wykrycia gruczolaków kolonoskopia jest wykonywana co roku aż do planowej kolektomii[122].
Badania przesiewowe w kierunku innych nowotworów niż rak jelita grubego są przeprowadzane od momentu rozpoznania polipów lub od 25–30. roku życia. Gastroskopia powinna być przeprowadzana co 5 lat. Niektórzy autorzy sugerują wykonywanie kontrastowego badania radiologicznego lub endoskopii kapsułkowej. Konieczne jest coroczne wykonywanie USG tarczycy i szyjki macicy u kobiet. Zaleca się wykonywanie TK lub MRI jamy brzusznej u chorych z wywiadem guza desmoidalnego. Zaleca się wykonanie profilaktycznej kolektomii z zachowaniem zwieraczy odbytu. Uzupełniająco mogą być stosowane niesteroidowe leki przeciwzapalne: sulindak i celekoksyb[122].
Chemioprofilaktyka raka jelita grubego
- Niesteroidowe leki przeciwzapalne
Kilka badań wskazuje, że regularne przyjmowanie kwasu acetylosalicylowego (aspiryna) przez pięć lat w dawce 300 mg dziennie zmniejsza ryzyko rozwoju łagodnych gruczolaków i ryzyko rozwoju na ich bazie raka jelita grubego, które jest mniejsze po 10-letnim stosowaniu leków[145][146][147]. Związek kwasu acetylosalicylowego z zapobieganiem raka jest mniejszy jeśli jest stosowany w dawce niższej niż 300 mg dziennie[148]. Selektywne inhibitory COX-2 (koksyby) celekoksyb i rofekoksyb zmniejszają ryzyko nawrotu gruczolaków[149][150][151], jednak ich użycie wiąże się ze znacznym wzrostem ryzyka sercowo-naczyniowego[152][153]. Sulindak, niesteroidowy lek przeciwzapalny, zmniejsza ilość i wielkość polipów w zespole rodzinnej polipowatości gruczolakowatej[154][155].
Kwas acetylosalicylowy i inhibitory COX redukują ryzyko zachorowania na raka jelita grubego, jednak ich stosowanie w populacji ogólnej nie jest uzasadnione i jest niezalecane, ze względu na ryzyko niekorzystnego działania w postaci zwiększonego ryzyka udaru mózgu i krwawienia z przewodu pokarmowego w przypadku aspiryny i zwiększonego ryzyka sercowo-naczyniowego w przypadku koksybów, które przeważają nad ewentualnymi korzyściami[135]. W niektórych niewielkich populacjach o szczególnie wysokim ryzyku raka korzyści z takiej profilaktyki przeważają nad ryzykiem związanym z działaniami niepożądanymi[135][138].
- Statyny
Wyniki metaanalizy nie potwierdzają by statyny stosowane w leczeniu hipercholesterolemii zmniejszały ryzyko rozwoju raka jelita grubego[156][138].
Epidemiologia

<2,5 2,5–5 5–7,5 7,5–10 | 10–12,5 12,5–15 15–17,5 17,5–20 | 20–22,5 22,5–25 25–27,5 >27,5 |
brak danych

Rak jelita grubego jest jednym z najczęściej rozpoznawanych nowotworów. Pod względem zapadalności na świecie u kobiet stanowi drugi po raku piersi nowotwór złośliwy, a u mężczyzn trzeci po raku płuca i raku prostaty[3][4]. Stanowi aż 10% wszystkich nowotworów złośliwych u ludzi[4]. W 2012 roku na świecie rozpoznano 1 340 000 przypadków raka jelita grubego, co stanowiło około 10% wszystkich przypadków nowotworów złośliwych i 690 000 przypadków zgonów na świecie, co stanowi około 8% zgonów z powodu choroby nowotworowej[5][158][159]. W 2012 roku w Europie rozpoznano 450 000 przypadków raka jelita grubego i 215 000 przypadków zgonów[160][161]. W Stanach Zjednoczonych średnie ryzyko zachorowania w ciągu życia na raka jelita grubego ocenia się na 5% dla mężczyzn i 4,7% dla kobiet[162].
Zapadalność na świecie jest znacznie zróżnicowana i występują ponad dziesięciokrotne różnice w częstości zachorowań. Większość przypadków występuje w krajach rozwiniętych[28]. Najwyższą zapadalność sięgającą 40 przypadków na 100 000 obserwuje się w Europie Zachodniej, Stanach Zjednoczonych, Australii i Nowej Zelandii, podczas gdy najniższą zapadalność stwierdza się w Azji, Afryce, Ameryce Południowej[28][159].
W Europie Zachodniej, Północnej i Stanach Zjednoczonych obserwuje się stabilizację częstości występowania. Z kolei w Europie Środkowej i Wschodniej, niektórych krajach Azjatyckich obserwuje się wzrost zapadalności na raka jelita grubego, co jest związane ze wzrostem zamożności tych społeczeństw[28].
Zdecydowana większość (90–95% przypadków) zachorowań pojawia się po 50. roku życia[159][28]. Średni wiek w momencie rozpoznania wynosi 64 lata[163]. Na świecie częstość występowania raka jelita grubego u kobiet i mężczyzn jest porównywalna[163], choć choroba częściej dotyczy mężczyzn[164]. U kobiet częściej obserwuje się prawostronne występowanie raka jelita grubego[164][165]. Kobiety osiągają lepsze przeżycia całkowite niż mężczyźni[163]. Większe ryzyko zachorowania na raka jelita grubego dotyczy osób rasy czarnej[162].
Polska jest krajem o średniowysokiej częstości zachorowań. Zapadalność u mężczyzn wynosi 30 przypadków na 100 000, a u kobiet 18 przypadków na 100 000[159]. W Polsce w 2010 roku rozpoznano 15600 przypadków raka jelita grubego, z czego 8700 przypadków u mężczyzn i 7100 przypadków u kobiet[159]. Obserwuje się tendencję wzrostową zapadalności i również umieralności na raka jelita grubego[137][166][167]. Od 1980 roku stwierdzono czterokrotny wzrost zachorowań u mężczyzn i trzykrotny wzrost u kobiet[159][137].
Histopatologia

.jpg)



.jpg)
.jpg)
Raki jelita grubego w klasyfikacji WHO z 2010 roku są dzielone na[168][169]:
- gruczolakorak (ang. adenocarcinoma, ICD-O 8140/3),
- śluzowy (ang. mucinous adenocarcinoma, ICD-O 8480/3),
- sygnetowaty (ang. signet ring cell carcinoma, ICD-O 8490/3),
- rdzeniasty (ang. medullary carcinoma, ICD-O 8510/3),
- sitowaty typu czopiastego (ang. cribriform comedo-type adenocarcinoma, ICD-O 8201/3),
- drobnobrodawkowaty (ang. micropapillary carcinoma, ICD-O 8265/3),
- ząbkowany (ang. serrated adenocarcinoma, ICD-O 8213/3),
- rak płaskonabłonkowy (ang. squamous cell carcinoma, ICD-O 8070/3),
- rak gruczołowopłaskonabłonkowy (ang. adenosquamous carcinoma, ICD-O 8560/3),
- rak niezróżnicowany (ang. undifferentiated carcinoma, ICD-O 8020/3),
- rak wrzecionowatokomórkowy (ang. spindle cell carcinoma, ICD-O 8032/3),
- rak neuroendokrynny o wysokiej złośliwości (ang. high grade neuroendocrine carcinoma, ICD-O 8246/3),
- wielkokomórkowy (ang. large cell, ICD-O 8013/3),
- drobnokomórkowy (ang. small cell, ICD-O 8041/3).
Gruczolakorak stanowi 90–95% wszystkich nowotworów złośliwych jelita grubego u człowieka[170][171][172][173][174]. Zdecydowana większość guzów jest zlokalizowana w esicy i odbytnicy[175]. Najczęściej jest zlokalizowany w odbytnicy (30–50% przypadków), esicy (15–20%), następnie we wstępnicy (14%), poprzecznicy (9%) i zstępnicy (6%)[15]. Obserwuje się wzrost częstości zachorowań na raka położonego po prawej połowie jelita grubego i zmniejszenie częstości zachorowań na raka dystalnej części jelita grubego[176][177]. U osób poniżej 40. roku życia[178], u osób starszych[179][177] oraz u kobiet częściej rozpoznaje się raka prawej połowy jelita grubego[164][165][162]. W 3,5% przypadków w momencie rozpoznania stwierdza się drugi synchroniczny rak jelita grubego[180].
Makroskopowo guzy przyjmują różne formy morfologiczne. Guzy bliższej części okrężnicy wykazują tendencję do egzofitycznego/grzybiastego wzorca wzrostu z tworzeniem dużej polipowatej masy, często z owrzodzeniem. W dalszej części okrężnicy guzy wykazują tendencje do płaskiego, pierścieniowatego wzrostu prowadzącego do zwężenia światła jelita. Brzeg nacieku nowotworowego jest obwałowany. Guzy mogą być uszypułowane lub siedzące[181][182][183].
Makroskopowo rak jelita grubego przyjmuje formy (klasyfikacja endoskopowa)[184][185]:
- postać polipowata – egzofityczna masa wewnątrz światła jelita bez owrzodzenia,
- postać grzybiasta (wrzodziejąco-polipowata) – polipowata masa z wyraźnym owrzodzeniem,
- postać wrzodziejąca – powierzchnia zmiany jest owrzodziała, owrzodzenie sięga poniżej poziomu prawidłowej błony śluzowej,
- postać rozlana (linitis plastica) – rak jednolicie naciekający ścianę jelita, często prowadzący do jego zwężenia.
- Gruczolakorak
Typowo gruczolakoraki jelita grubego tworzą średniej wielkości struktury gruczołowe, zmienność wielkości, wzoru oraz ilość zrębu jest umiarkowana. Atypowe komórki wysokozróżnicowanego gruczolakoraka są walcowate, a w miarę spadku stopnia zróżnicowania komórki przyjmują kształt sześcienny. W świetle gruczołów mogą występować mucyny, komórki albo same jądra komórkowe. Figury mitotyczne zwykle są liczne[186].
Rak dobrze zróżnicowany w 95% zawiera gruczoły (stopień złośliwości histologicznej G1), rak średnio zróżnicowany zawiera 50–95% struktury gruczołowe (stopień złośliwości G2), rak słabo zróżnicowany zawiera 50–5% struktur gruczołowych (stopień złośliwości G3), a rak niezróżnicowany poniżej 5% struktur gruczołowych (stopień złośliwości G4)[187][170]. W systemie dwustopniowym niski stopień złośliwości obejmuje stopnie G1 i G2, a wysoki stopień złośliwości G3 i G4[169]. W 70% przypadków gruczolakorak jelita grubego wykazuje średni stopień zróżnicowania, dobrze zróżnicowane gruczolaki stanowią 10% guzów, a nisko zróżnicowane 20% przypadków[170][186].
- Gruczolakorak śluzowy
Jest to podtyp gruczolakoraka o znacznej zawartości mucyn, których zawartość przekracza 50%. Stanowi około 10% wszystkich raków jelita grubego u ludzi[188]. Makroskopowo jest miękkim, galaretowatym guzem[188]. Mikroskopowo nowotwór zawiera duże struktury gruczołowe z dużymi pozakomórkowymi polami mucyny. Mogą występować pasma komórek lub pojedyncze komórki. Często stwierdza się niestabilność mikrosatelitarną[189][170]. Może towarzyszyć zespołowi Lyncha[188].
- Gruczolakorak sygnetowaty
Jest to rzadki podtyp gruczolakoraka, stanowi tylko poniżej 1% przypadków raka jelita grubego u ludzi[170][190]. 30% przypadków gruczolakoraka sygnetowatego dotyczy chorych na wrzodziejące zapalenie jelita grubego[190]. Nowotwór zwykle wykazuje wrzodziejący lub naciekający typ wzrostu[190]. Gruczolakorak sygnetowaty zawiera przynajmniej 50% komórek sygnetowatych, które cechują się obecnością dużej wakuoli zawierającej mucyny wypierającej jądro komórkowe na obwód komórki. Komórki mogą wykazywać rozlany, naciekający wzorzec wzrostu z minimalną ilością zewnątrzkomórkowej mucyny lub być obecne w dużych zewnątrzkomórkowych polach mucyny. Jest to nowotwór o słabym zróżnicowaniu i wysokim stopniu złośliwości[189][170].
- Gruczolakorak rdzeniasty
Jest to bardzo rzadki podtyp gruczolakoraka. Nowotwór wykazuje lity wzór utkania, jest zbudowany ze sznurów okrągłych komórek z pęcherzykowatym jądrem, wyraźnymi jąderkami i obfitą cytoplazmą[191][170]. Zmienność wielkości i kształtu jąder komórkowych jest niewielka[192]. Charakterystyczny jest naciek limfocytarny. Jest mocno związany z niestabilnością mikrosatelitarną[191][170]. Mimo niskiego zróżnicowania zwykle charakteryzuje go dobre rokowanie[170].
- Rak gruczołowopłaskonabłonkowy
Jest to bardzo rzadki typ raka wykazujący cechy gruczolakoraka i raka płaskonabłonkowego, stanowi około 0,06% przypadków raka jelita grubego u ludzi[193]. W obrębie guza mogą występować odrębne obszary z występowaniem przewagi gruczolakoraka lub raka płaskonabłonkowego albo oba typy są wymieszane[189].
- Rak płaskonabłonkowy
Czysty rak płaskonabłonkowy jest bardzo rzadką postacią raka jelita grubego[189]. W nowotworze nie mogą występować ogniska raka gruczołowego i musi zostać wykluczony rak odbytu[192].
- Rak wrzecionowatokomórkowy
Jest to bardzo rzadka postać raka jelita grubego. Cechuje się obecnością elementów nabłonkowych i mezynchymalnych[194].
- Rak niezróżnicowany
Jest to rzadki podtyp raka jelita grubego wykazujący cechy różnicowania w kierunku raka, ale bez tworzenia struktur gruczołowych ani innych dowodów na różnicowanie[190]. Wykazuje znaczną zmienność cech histologicznych[191]. Nowotwór wykazuje znaczną komórkowość i dużą skłonność do rozległej martwicy[190].
Zmiany przednowotworowe


.jpg)


.jpg)

Zmiany przednowotworowe są ściśle związane z karcynogenezą raka jelita grubego. Istnieją dwie główne ścieżki karcynogenezy prowadzące do raka jelita grubego: ścieżka związana z niestabilnością chromosomalną związaną z mutacją genu APC oraz ścieżka związana z mutacją genów mutatorowych, głównie hMSH2 i hMLH1, prowadząca do niestabilności genetycznej. Zmiany przednowotworowe są ściśle powiązane z tymi ścieżkami karcynogenezy. Gruczolak jest łagodną zmianą nabłonkową powiązaną z mutacją APC będącą obserwowalnym prekursorem raka inwazyjnego. Sekwencja gruczolak-rak jest główną drogą prowadzącą do raka, obserwowaną w 85% przypadków raka jelita grubego u ludzi. Z kolei zmiany ząbkowane są powiązane ze ścieżką mutacji genów mutatorowych i około 15% przypadków raka jelita grubego jest następstwem tej drogi karcynogenezy[195][196]. Makroskopowo łagodne gruczolaki mogą tworzyć uniesione zmiany nazywane polipem lub zmiany nieuniesione (płaskie)[197].
Zmiany przednowotworowe są stwierdzane u około 25% osób z populacji ogólnej poddawanych kolonoskopii. Częstość występowania rośnie wraz z wiekiem i u osób po 80. roku życia łagodne zmiany przednowotworowe mogą być obecne u 85% osób poddanych kolonoskopii. Dwie trzecie polipów jest umiejscowiona po lewej połowie jelita grubego[198]. U 2% bezobjawowych osób poddanych kolonoskopii stwierdza się zmiany o wysokim stopniu neoplazji i u 1% badanych rozpoznaje się raka inwazyjnego[198].
Niezłośliwe zmiany nabłonkowe są klasyfikowane jako[199][200]:
- gruczolaki konwencjonalne,
- gruczolak cewkowy,
- gruczolak cewkowo-kosmkowy,
- gruczolak kosmkowy,
- zmiany ząbkowane,
- polip hiperplastyczny,
- gruczolak/polip ząbkowany siedzący,
- tradycyjny gruczolak ząbkowany,
- zmiany mieszane.
Ogniska nieprawidłowych krypt
Ognisko nieprawidłowych krypt (ang. aberrant crypt focus) jest najwcześniejszym morfologicznym prekursorem neoplazji śródnabłonkowej i poprzedza rozwój polipów nabłonkowych. Zmiany te nie są widoczne w rutynowej endoskopii. W badaniu mikroskopowym lub endoskopii powiększającej są widoczne jako kilka do dwustu powiększonych krypt o poszerzonym ujściu, otoczonych pogrubiałym nabłonkiem i o zmniejszonej ilości śluzu. Progresja ognisk nieprawidłowych krypt do gruczolaka jest elementem karcynogenezy[197][196].
Gruczolaki jelita grubego
Gruczolaki jelita grubego są to łagodne zmiany nowotworowe z zaznaczonymi cechami neoplazji śródnabłonkowej. W obrazie histopatologicznym występują zmiany cytologiczne i architektoniczne. Zmiany cytologiczne obejmują wydłużenie i nawarstwienie jąder komórkowych, powiększenie jąder komórkowych, hiperchromazja i heterochromazja chromatyny, utratę polarności komórki, zwiększenie stosunku jądrowo-cytoplazmatycznego oraz obecność licznych figur podziału. Na zmiany architektoniczne składają się rozrost i stłoczenie krypt oraz wytworzenie kosmków[201]. Makroskopowo gruczolaki mogą być zmianami uniesionymi, płaskimi lub zapadniętymi (wrzód)[197].
Mikroskopowo na podstawie udziału elementu kosmkowego gruczolaki klasyfikuje się jako gruczolaki cewkowe (ang. tubular adenoma), gruczolaki cewkowo-kosmkowe (ang. tubulo-villous adenoma) i gruczolaki kosmkowe (ang. villous adenoma). Gruczolak cewkowy musi być zbudowany przynajmniej w 80% z elementów cewkowych, a gruczolak kosmkowy w przynajmniej 80% z elementów kosmkowych. Gruczolaki bez takiej przewagi jednej grupy elementów klasyfikowane są jako gruczolaki cewkowo-kosmkowe[196].
Gruczolak cewkowy jest zbudowany z przylegających, regularnych lub rozgałęzionych cew gruczołowych. Gruczolak kosmkowy jest zwykle zmianą siedzącą o kosmatej powierzchni zbudowanej z wydłużonych struktur przekraczających dwukrotnie grubość prawidłowej błony śluzowej[196][202].
Ze względu na nasilenie stopnia zmian cytologicznych i architektonicznych gruczolaków wyróżnia się neoplazję (dysplazja) niskiego i wysokiego stopnia[201]. Neoplazja wysokiego stopnia cytologicznie cechuje się obecnością nasilonej atypii z obecnością zaokrąglonych jąder komórkowych, pogrubieniem chromatyny, obecnością wyraźnych jąderek i utratą polarności komórki. W architekturze zmiany stwierdza się stłoczenie struktur gruczołowych z wytworzeniem struktur sitowatych[201][170].
Największe ryzyko transformacji do raka inwazyjnego charakteryzuje zmiany z obecną neoplazją wysokiego stopnia, zmiany o wielkości >10 mm lub obecność >20% elementu kosmkowego (polipy cewkowo-kosmkowe i kosmkowe)[201].
Zmiany ząbkowane
Zmiany ząbkowane stanowią heterogenną grupę zmian wykazujących ząbkowaną architekturę przekroju gruczołu, związaną z nadmiernym pofałdowaniem nadmiernie rozrośniętego nabłonka gruczołowego[199][170].
- Polip hiperplastyczny
Polipy hiperplastyczne stanowią zdecydowaną większość zmian ząbkowanych. Są to niewielkie zmiany o wielkości około 5 mm, rzadko przekraczające wielkość 10 mm. Zwykle są położone w lewej połowie jelita grubego. Makroskopowo polip hiperplastyczny jest trudny do odróżnienia od gruczolaków. Mikroskopowo budują go podłużne, proste krypty zawierające w świetle ząbkowanie tworzone przez nadmiernie rozrośnięte komórki nabłonka. Strefa proliferacyjna jest ograniczona do części podstawnej krypt, która pozostaje wąska i nie zawiera ząbkowania[170]. Nie stwierdza się dysplazji[199].
- Gruczolak ząbkowany siedzący i polip ząbkowany siedzący
Pojęcia gruczolak ząbkowany siedzący i polip ząbkowany siedzący są stosowane zamiennie[170]. Stanowią około 1–9% polipowatych zmian jelita grubego[198]. Mikroskopowo cechuje się obecnością cech dysplazji. Występuje nadmierne ząbkowanie, które występuje na całej długości krypty, poszerzenie krypty i obecność rozgałęzień krypt. W przeciwieństwie do polipów hiperplastycznych gruczolak/polip ząbkowany siedzący stanowi zmianę przednowotworową[170].
- Tradycyjny gruczolak ząbkowany
Tradycyjny gruczolak ząbkowany jest stosunkowo rzadką zmianą, stanowi około 1% zmian polipowatych jelita grubego. Mikroskopowo również należy do zmian ząbkowanych, ale prezentuje obecność cech utkania cewko-kosmkowego lub kosmkowego[198][200]. Charakteryzuje się neoplazją nabłonka niskiego stopnia, rzadziej wysokiego stopnia, z ząbkowaniem nabłonka[170]. Występują wydłużone jądra komórkowe, pseudonawartwienia jąder i eozynofilna cytoplazma[203]. Typowa jest obecność krypt ektopowych[200]. Zwykle występują po lewej połowie jelita grubego[203].
Patogeneza

Karcynogeneza raka jelita grubego nie jest całkowicie poznana. Istotą tego procesu jest powstanie kaskady zmian cytogenetycznych i epigenetycznych w rezultacie długiej ekspozycji na środowiskowe i wrodzone czynniki ryzyka raka zaburzające homeostazę komórkową[204]. Karcynogeneza jest procesem wieloetapowym. Pojedyncza mutacja nie jest wystarczająca do rozwoju raka, a choroba jest efektem wielu kolejnych mutacji[205][206]. Kolejne mutacje pojawiają się w różnym czasie w różnych miejscach materiału genetycznego[207]. Mutacje prowadzą do wyłączenia niektórych genów lub przeciwnie do wzmocnienia ich transkrypcji[208].
Karcynogeneza raka jelita grubego może być podzielona na inicjację, promocję i progresję[205]. W najwcześniejszym etapie inicjacji dochodzi do pierwszej zmiany genetycznej. Jeśli mutacja wykazuje biologiczne znaczenie i komórka jest zdolna do przetrwania może otwierać to drogę do dalszych zmian genetycznych w etapie progresji. Zmiany genetyczne i epigenetyczne prowadzą do powstania klonu komórek o przewadze wzrostowej nad innymi komórkami[206]. Dochodzi do gromadzenia komórek, które nabywają kolejne mutacje. W miarę kolejnych mutacji genetycznych komórki nabywają kolejnych cech komórek nowotworowych, w tym zdolność do inwazji, migracji i przerzutowania[208]. Proces karcynogenezy jest rozciągnięty w czasie, od pierwszej mutacji do rozwoju raka inwazyjnego mija około 10–20 lat[209][208] i około 5–10 lat od pojawienia się polipa do raka inwazyjnego[210].
Istnieją dwie główne ścieżki prowadzące do raka jelita grubego. Najczęstszą z nich jest szlak gruczolak–rak, w którym obserwuje się progresję zmian morfologicznych od łagodnego gruczolaka, który następnie ulega progresji przez neoplazję wysokiego stopnia do raka inwazyjnego. Karcynogenezę w tym szlaku rozpoczyna mutacja genu supresorowego APC, a następnie pojawiają się mutacje protoonkogenów i innych genów supresorowych ostatecznie doprowadzających do niestabilności chromosomalnej. W mniej częstym modelu zmian ząbkowanych pierwszym zjawiskiem jest wyłączenie genów naprawiających materiał genetyczny, co prowadzi do zjawiska niestabilności mikrosatelitarnej. Dopiero w kolejnym etapie dochodzi do aktywacji onkogenów[195][211].
Rola czynników środowiskowych
- Czynniki dietetyczne
Nieprawidłowa dieta odgrywa ważną rolę w karcynogenezie raka jelita grubego. Do transformacji nowotworowej dochodzi w następstwie długotrwałej ekspozycji na czynniki rakotwórcze[212]. Nadmierne spożywanie czerwonego mięsa jest związane ze zwiększonym ryzykiem rozwoju raka jelita grubego. Prawdopodobnie karcynogeneza związana z czerwonym mięsem jest efektem powstawania związków N-nitrozowych i metabolizmu zwierzęcego hemu[213].
Wiele związków N-nitrozowych, w tym nitrozoaminy i nitrozoamidy jest związkami rakotwórczymi w karcynogenezie licznych nowotworów. Mogą one reagować z materiałem genetycznym komórki sprzyjając powstawaniu mutacji genetycznych[214]. Obecność związków N-nitrozowych stwierdzono w przetworzonym mięsie, wędzonych rybach i serze oraz piwie[215][214].
Związki N-nitrozowe w znacznej części powstają endogennie w rezultacie nadmiernej ekspozycji na czerwone mięso[214]. Azotany, obecne w żywności jako środki konserwujące, w wyniku metabolizmu obecnych w przewodzie pokarmowym bakterii ulegają redukcji do azotynów, które mogą reagować z aminami i amidami dając rakotwórcze związki N-nitrozowe[216]. Istnieje zależność dawki czerwonego mięsa i ilości endogennych związków N-nitrozowych, ale nie stwierdzono jej dla białego mięsa[217][213]. Różnica tłumaczona jest większą zawartością hemu w czerwonym mięsie niż w białym mięsie[218][213]. Hem katalizuje reakcje powstawania związków N-nitrozowych niezależnie od wytwarzania tych związków przez bakterie[218][214]. Hem może wykazywać działanie rakotwórcze poprzez wywoływanie uszkodzenia nabłonka i stymulacji do jego naprawy[219] oraz peroksydację tłuszczów i wytwarzanie lipoperoksydów[220].
Prawdopodobnie w patogenezie raka jelita grubego pewną rolę pełnią policykliczne węglowodory aromatyczne i heterocykliczne aminy aromatyczne. Heterocykliczne aminy aromatyczne powstają podczas obróbki mięsa w wysokiej temperaturze, w tym podczas smażenia, pieczenia i grillowania. Z kolei głównym źródłem policyklicznych węglowodorów aromatycznych są gotowane, wędzone i grillowane mięso i ryby[214]. Ryzyko raka jelita grubego jest bardziej związane ze sposobem gotowania mięsa niż samym przyjmowaniem wielopierścieniowych węglowodorów aromatycznych i heterocyklicznych amin[221][214].
Wysokie spożycie tłuszczów wpływa na karcynogenezę prawdopodobnie poprzez zwiększenie wydzielania żółci, która w przewodzie pokarmowym przy udziale bakterii ulega przemianie do wtórnych kwasów żółciowych, które promują karcynogenezę[214].
- Nieswoiste zapalenia jelit
Karcynogeneza w nieswoistych zapaleniach jelit jest związana z nasileniem stresu oksydacyjnego, który przyczynia się do uszkodzenia DNA, oraz wpływu hamującego apoptozę i nasilającego przeżycie komórki, a także utworzenia mikrośrodowiska promującego wzrost, migrację komórek oraz neoangiogenezę[222]. Przewlekłe zapalenie nasila wymianę komórek błony śluzowej jelita grubego, a proces jest szczególnie nasilony w miejscach aktywnego zapalenia. Prawdopodobnie cytokiny i czynniki wzrostu związane z zapaleniem mogą stymulować karcynogenezę[223]. TNF-α może powodować uszkodzenie DNA poprzez generację stresu oksydacyjnego[224][223]. Czynnik NF-κB reguluje ekspresje cytokin prozapalnych i nasilenie poziomu reakcji zapalnej. Niektóre geny kontrolowane przez ten czynnik transkrypcyjny wykazują działanie hamujące apoptozę, zwiększające przeżycie komórki i regulujące różnicowanie się komórki[225][223].
- Palenie tytoniu
Palenie tytoniu dostarcza wiele substancji rakotwórczych, w tym wielopierścieniowe węglowodory aromatyczne, aminy aromatyczne, nitrozoaminy i aminy heterocykliczne, które ulegają metabolizmowi do związków zdolnych do połączenia z DNA, co wywołuje naprawę DNA, w tym poprzez wycięcie. Niektóre karcynogeny obecne w dymie tytoniowym (NNK: 4-(metylonitrozoamino)-1-(3-pirydylo)-1-butanon) wykazują powinowactwo do receptorów nikotynowych ostatecznie wpływając na powstawanie wolnych rodników tlenowych i stresu oksydacyjnego. Z kolei stymulacja receptorów β-adrenergicznych wywołuje szereg reakcji metabolicznych i immunomodulujących[226].
Szlak gruczolak–rak
Jest to główna sekwencja mutacji prowadząca do raka jelita grubego, dotyczy ona 80% przypadków sporadycznego raka i wszystkie przypadki rodzinnej polipowatości gruczolakowatej. W tym modelu sekwencja odpowiednich zmian morfologicznych z powiązanymi mutacjami prowadzi ostatecznie do raka[227]. Akumulacja zmian cytogenetycznych dających przewagę wzrostową nad innymi komórkami prowadzi do progresji zmiany stopnia neoplazji (dysplazji) od gruczolaka do raka inwazyjnego[228]. Sekwencja zmian jest inicjowana przez mutację genu APC, następnie pojawiają się mutacje KRAS (k-RAS), p53 i DCC[227]. Ważną rolę pełnią mutacje genów naprawiających DNA: MLH1, MSH2 i MSH6[210]. Mutacja APC za pośrednictwem szlaku Wnt/β-katenina oraz innych genów supresorowych prowadzi do zaburzenia kontroli apoptozy i kontroli cyklu komórkowego, co napędza proliferację komórek nowotworowych[195]. Ostatecznie mutacje genów supresorowych, onkogenów i genów naprawiających DNA (mutatorowych) prowadzą do utraty stabilności genetycznej komórki, co umożliwia progresję zmiany do raka[229].
Szlak zmian ząbkowanych
Szlak zmian ząbkowanych prowadzi do raka poprzez mechanizm zaburzenia genów mutatorowych odpowiedzialnych za naprawę materiału genetycznego komórki, co powoduje niestabilność mikrosatelitarną (MSI)[195]. Pierwszymi mutacjami w szlaku zmian ząbkowanych są mutacje protoonkogenów KRAS lub BRAF. Zarówno mutacje KRAS, jak i BRAF są odpowiedzialne za aktywacje szlaku MAPK. Wywołuje to niekontrolowaną proliferację komórek, zwiększenie ich zdolności do przeżycia (unikanie apoptozy) i zdolność do migracji komórki[230]. Kolejną zmianą jest powstanie niestabilności mikrosatelitarnej spowodowanej wyłączeniem genów mutatorowych. Prowadzi to do wrażliwości na akumulację zmian genetycznych w genach zlokalizowanych w regionach mikrosatelitarnych[231][230]. W zmianach ząbkowanych często obserwuje się fenotyp metylatora wysp CpG (ang. CpG island methylator phenotype, CIMP). Metylacja wysp CpG hamuje ekspresję wielu genów, w tym genów supresorowych takich jak MLH1[230][232][233].
Podłoże cytogenetyczne karcynogenezy raka jelita grubego
- APC i szlak Wnt/β-katenina
Gen APC pełni kluczową rolę w karcynogenezie raka jelita grubego. Jego mutacja determinuje wejście na szlak gruczolak–rak, którego rozwój bez tej mutacji jest mało prawdopodobny[212][234]. Mutacja genu APC lub mutacja genu β-kateniny jest stwierdzana w 80–90% przypadków tego raka, co sugeruje, że jest to wczesnym zdarzeniem w karcynogenezie raka jelita grubego[235][229][211][236].
Gen APC jest genem supresorowym blokującym przejście z fazy G1 do fazy S cyklu komórkowego. Główną funkcją białka APC jest regulacja szlaku Wnt/β-katenina[237][212]. Szlak Wnt/β-katenina uczestniczy w regulacji proliferacji, apoptozy i różnicowania się komórki[227]. W prawidłowych warunkach APC reguluje szlak Wnt poprzez wiązanie się z β-kateniną i obniżając jej stężenie. W przypadku wystąpienia mutacji białko APC nie posiada zdolności z wiązania β-kateniny i stężenie niezwiązanej β-kateniny wzrasta, co pozwala jej wejść do jądra komórkowego i stale aktywować geny odpowiedzialne za podział komórki, co nasila proliferację komórki i upośledza jej różnicowanie[227][212]. β-katenina jest odpowiedzialna za migrację komórek. Jej nagromadzenie się w wyniku mutacji genu APC zaburza migrację komórek w kierunku powierzchni błony śluzowej, gdzie po kilku dniach uległaby usunięciu. W rezultacie akumulacja niezróżnicowanych komórek w kryptach jelitowych prowadzi do tworzenia się polipów[229]. Białko APC za pośrednictwem β-kateniny i E-kadheryny kontroluje adhezje komórek oraz migrację komórek poprzez wpływ na mikrotubule. Białko APC poprzez oddziaływanie na aktynę wpływa na cytoszkielet[227].
- p53
p53 jest ważnym genem supresorowym i pełni istotną rolę w utrzymaniu stabilności genomu. W przypadku obecności błędów podczas replikacji białko p53 posiada zdolność zatrzymania cyklu komórkowego i wpływa na naprawę materiału genetycznego. Gdy uszkodzenia nie udaje się naprawić kieruje komórkę na szlak apoptozy[212].
p53 może wywołać apoptozę drogą wewnątrzpochodną i zewnątrzpochodną za pośrednictwem Fas. p53 posiada zdolność do wywołania ekspresji białek Bax, Noxa i PUMA, które obniżają stężenie antyapoptycznego białka Bcl-2, które ma za zadanie obniżać stężenie cytochromu C AIF. Uwolniony cytochrom C aktywuje kaspazy, które kontrolują szlak apoptozy. p53 reguluje w górę ekspresję Fas i DR5 (TRAIL-R2), które mogą wywołać apoptozę drogą zewnątrzpochodną. p53 w przypadku rozpoznania uszkodzenia DNA promuje powstawanie białka p21, które wiąże kompleks CDK4/CDK6, CDK2 i CDK1, co ostatecznie blokuje różne geny i zatrzymuje cykl komórkowy[238].
Mutacja powodująca unieczynnienie genu p53 jest kluczowym etapem w karcynogenezie raka jelita grubego. Odgrywa ważną rolę w przejściu gruczolaka do raka[238]. Mutacja p53 jest stwierdzana w 34% przypadków raka w prawej połowie jelita grubego i 45% przypadków raka po lewej połowie okrężnicy[239][238].
- DCC (utrata heterozygotyczności 18q)
W zaawansowanym raku jelita grubego często dochodzi do utraty jednej kopii genów w regionie 18q21 zawierającego gen DCC. W 70% przypadków raka jelita grubego stwierdza się utratę heterozygotyczności obejmującą region genu DCC[240][241]. Zwykle druga kopia genu jest dotknięta mutacją[229].
Gen DCC koduje przezbłonowe białko DCC wykazujące właściwości antyonkogenu. DCC blokuje wzrost komórki w przypadku nieobecności ligandu NTN1 (netrin-1), który jest produkowany w kryptach jelitowych i odpowiada za prawidłową regulację wzrostu, różnicowania się i migracji komórek[229]. W przypadku nieobecności ligandu, DCC indukuje aktywacje kaspazy 9 oraz kaspazy 3 prowadząc do apoptozy[241][242]. Z drugiej strony, w przypadku konstytutywnej aktywacji NTN1, DCC może promować wzrost komórek nowotworowych[241]. Mutacja genu DCC powoduje brak wiązania z NTN1 i nieprawidłowe nadmierne przeżycie komórek nowotworowych[229].
- EGFR
Receptor naskórkowego czynnika wzrostu (EGFR) jest przezbłonową glikoproteiną o aktywności kinazy tyrozynowej. Głównym ligandami są naskórkowy czynnik wzrostu (EGF) i TGF-α, który powoduje dimeryzację receptora i transdukcję sygnału poprzez aktywacje szlaków MAPK/ERK i PI3K/AKT/mTOR[229][243]. Zwiększa to zdolność do proliferacji, chroni komórki przed apoptozą, ułatwia migrację komórek i promuje angiogenezę[229][212]. Zwiększoną ekspresję EGFR stwierdza się w 25–80% przypadków raka jelita grubego[244].
- KRAS
Białko KRAS (k-RAS) jest protoonkogenem, należy do nadrodziny białek RAS. Białko ma za zadanie transdukcję sygnału wzrostowego, głównie z układu EGFR[245]. Mutacja onkogenu KRAS występuje w 30–50% przypadków raka jelita grubego[227][245][246][247][248][249].
KRAS jest niewielką GTP-azą odpowiadającą za rozkład GTP do GDP. Aktywacja KRAS jest promowana przez białka GEF (guanine nucleotide exchange factor), a inaktywacji pośredniczy GAP (GTPase-activating proteins)[250][251]. Zaktywowane białko KRAS poprzez białka pośredniczące Ras, RAF i MEK wpływa na szlaki MAPK/ERK i PI3K/AKT/mTOR[227].
Mutacja aktywująca KRAS zaburza interakcje KRAS z hamującym jej działanie GAP, co doprowadza do konstytutywnej aktywności KRAS i stałego pobudzania zależnych od nich szlaków[245]. Prowadzi to do stymulacji wzrostu, proliferacji, oporności na apoptozę[229][252][245].
- BRAF
BRAF jest kinazą seroninową-treoninową aktywującą kinazy MAP/ERK należącą do rodziny kinaz RAF. Gen BRAF jest zaliczany do protoonkogenów[229]. Gen wpływa na proliferację komórek, dojrzewanie i apoptozę[227]. Mutacja aktywująca BRAF jest stwierdzana w 10% przypadków raka jelita grubego[229][253], w 90% dotyczy ona kodonu V600E[254][227]. W wyniku mutacji dochodzi do konstytutywnej aktywacji BRAF, co prowadzi do promowania nadmiernej proliferacji komórek niezależnej od czynników wzrostowych i oporności komórek na apoptozę[255][227].
- Geny mutatorowe (MMR) i niestabilność mikrosatelitarna (MSI)
Geny mutatorowe kodują białka odpowiedzialne za korekcję błędów powstających spontanicznie podczas replikacji materiału genetycznego. Korekcja ta polega na usuwaniu źle sparowanych nukleotydów lub powstających pętli (insertion-deletion loop, IDL)[256]. Pętle IDL powstają w wyniku poślizgu polimerazy DNA podczas replikacji odcinków DNA z powtarzającymi się sekwencjami nukleotydów, w tym regionów mikrosatelitarnych. Jeśli białko naprawcze (np. kodowane przez gen mutatorowy) nie usunie błędnie wstawionego nukleotydu mutacja może się utrwalić, jeśli pętla IDL nie zostanie usunięta, może dojść do przesunięcia ramki odczytu[256]. Łatwym do zdiagnozowania efektem dysfunkcji genów mutatorowych jest wydłużanie lub skracanie regionu mikrosatelitów, określane jako niestabilność mikrosatelitarna[257][258]. Niestabilność mikrosatelitarna wysokiego stopnia (MSI-H) jest definiowana jako obecność niestabilności ≥30% badanych markerów, niestabilność mikrosatelitarna niskiego stopnia (MSI-L) definiuje się jako obecność niestabilności w 10–29% badanych markerach[229].
Do genów mutatorowych należą MLH1, MSH2, MSH6 i PMS2[229]. Wyłączenie genów mutatorowych odbywa się za pośrednictwem metylacji wysp CpG lub mutacji punktowych poszczególnych genów. W sporadycznym raku jelita grubego najczęściej dochodzi do wyłączenia genów MLH1 i PMS2. MLH1 zwykle jest wyłączany za pomocą hipermetylacji promotora genu[256]. Niestabilność mikrosatelitarną stwierdza się w 10–20% przypadków raka jelita grubego[259].
- VEGF
Czynnik wzrostu śródbłonka naczyniowego (VEGF) jest białkiem sygnalizacyjnym odpowiadającym za angiogenezę. Jest najważniejszym czynnikiem proangiogenetycznym w raku jelita grubego[260][261]. Nadekspresja VEGF jest obserwowana w 50–60% przypadków raka jelita grubego[261][262].
Wraz z rozwojem guza jego masa oraz aktywność metaboliczna przewyższają możliwości zaopatrzenia go w krew przez dotychczasowe unaczynienie i rozwija się niedokrwienie i niedotlenienie guza[212][262]. W konsekwencji obniżenia ciśnienia parcjalnego tlenu wydzielany jest HIF-1, który jest czynnikiem transkrypcyjnym przyczyniającym się do ekspresji VEGF. Czynnik HIF-1 współpracuje z IL-6 i TGF-β w ekspresji proangiogennej VEGF[227]. Wydzielanie VEFF jest również stymulowane przez onkogen KRAS[263]. VEGF bezpośrednio promuje tworzenie nowych naczyń krwionośnych[212]. VEGF jest kluczowym czynnikiem sprzyjającym progresji nowotworu[262].
Przebieg naturalny choroby
.jpg)
.jpg)

.jpg)
Większość przypadków raka jelita grubego rozwija się na bazie łagodnych zmian obserwowanych makroskopowo jako polipy, które wykazują różnice pod względem budowy histopatologicznej i niesionego ryzyka rozwoju raka[264][265]. Typ zmiany przednowotworowej jest odzwierciedleniem różnych szlaków karcynogenezy. Najważniejszą zmianą przednowotworową jest gruczolak. Zapoczątkowuje on szlak gruczolak–rak i na jego bazie rozwija się 80% przypadków raka[195]. Część raków powstaje na bazie zmian ząbkowanych, które również mogą tworzyć polipy[195]. Przejście z łagodnej zmiany do raka jest procesem wieloetapowym. Proces karcynogenezy od pierwszej mutacji do powstania raka trwa około 10–20 lat[209][208]. Tylko część gruczolaków ulega progresji do raka. Szacuje się, że łączne ryzyko powstania raka na bazie gruczolaka większego niż 1 cm po 5 latach wynosi 2,5%, po 10 latach 8% i po 20 latach 25%[266][267]. Rak początkowo dotyczy wyłącznie błony śluzowej, później dochodzi do inwazji warstwy mięśniowej[268]. Rak na wczesnym etapie rozwoju rośnie powoli, ale po inwazji warstwy podśluzowej szybkość wzrostu przyspiesza[269].
Miejscowo rak szerzy się poprzecznie poprzez kolejne coraz głębsze warstwy jelita oraz podłużnie w obrębie jednej z warstw narządu. Szerzenie się podłużne wzdłuż długości jelita rzadko daje ogniska raka dalsze niż 2 cm od guza pierwotnego[270]. Zaawansowany rak okrężnicy po spenetrowaniu surowicówki może szerzyć się poprzecznie na tkanki okołojelitowe i sąsiednie narządy. Rak w obrębie kątnicy i esicy może naciekać jajniki, jajowody, macicę i jelito cienkie. Rak poprzecznicy, zgięcia wątrobowego i śledzionowego może naciekać dwunastnicę, żołądek, trzustkę i śledzionę. Przednia ściana jamy brzusznej jest częściej zajęta w guzach położonych w wewnątrzotrzewnowych częściach jelita grubego, tylna ściana jest częściej zajęta w guzach jelita grubego w częściach pozaotrzewnowych[271][272]. Rak zlokalizowany w odbytnicy może szerzyć się na tkanki okołoodbytnicze i naciekać sąsiednie narządy w tym pęcherz moczowy i pochwę[181].
W późniejszym etapie nowotwór szerzy się za pośrednictwem naczyń chłonnych i krwionośnych dając przerzuty węzłowe i przerzuty odległe. Przerzuty drogą limfatyczną w okrężnicy zwykle nie pojawiają się do czasu inwazji do przydanki[273][272]. Penetracja przez nowotwór żył spływających do żyły wrotnej lub żyły głównej dolnej skutkuje pojawieniem się przerzutów odległych drogą krwionośną[189]. Możliwy jest naciek wzdłuż nerwów[274][275][276].
Przerzuty odległe zwykle pojawiają się w bardziej zaawansowanych stadiach choroby, jednak zdarza się ich obecność już we wczesnym etapie choroby[273][181]. Przerzuty, w związku z unaczynieniem wrotnym jelit, najczęściej lokalizują się w wątrobie. Najważniejszą pozawątrobową lokalizacją jest płuco, a następnie mózg i kości[277]. Przerzuty do lokalizacji pozawątrobowych częściej występują w raku odbytnicy niż okrężnicy, co jest związane z podwójnym unaczynieniem żylnym odbytnicy[277][268]. Obecność przerzutów w płucach wiąże się z większym ryzykiem stwierdzenia przerzutów w kościach i mózgu[277]. Przerzuty odległe są stwierdzane u około 35% chorych w momencie rozpoznania[270].
Większość guzów jest zlokalizowana w esicy i odbytnicy[175]. Guzy zlokalizowane w kątnicy i wstępnicy zwykle osiągają większe rozmiary niż guzy w pozostałych częściach jelita grubego. Guzy zlokalizowane w esicy i odbytnicy wykazują większą tendencję do zamykania światła jelita grubego i wywoływania niedrożności[171]. Z guzami po stronie prawej częściej współwystępują guzy metachroniczne[278].
U 7–30% chorych w następstwie wzrostu guza lub obecności przerzutów dochodzi do przepuszczającej lub całkowitej niedrożności mechanicznej jelit[279][280]. U około 2,5–10% przypadków w efekcie wzrostu guza może dojść do perforacji jelita grubego[281][282][283]. Perforacja prowadzi do powstania rozlanego zapalenia otrzewnej lub ropnia wewnątrzotrzewnowego[284]. Perforacja może być następstwem niedrożności jelit[285].
Rozpoznanie choroby






Podstawowym badaniem pozwalającym rozpoznać raka jelita grubego jest kolonoskopia[6]. Badanie umożliwia uwidocznienie guza oraz pobranie próbki do badania histopatologicznego. Wirtualna kolonoskopia TK lub MRI, endoskopia kapsułkowa oraz radiologiczne badanie dwukontrastowe pełnią pomocniczą rolę w rozpoznaniu i w tych badaniach stwierdzenie podejrzanej zmiany wymaga weryfikacji kolonoskopowej. W ocenie zaawansowania stosuje się tomografię komputerową, rezonans magnetyczny, ultrasonografię endoskopową oraz pozytonową tomografię emisyjną[8].
Endoskopia
Endoskopia jest podstawową metodą wykrywania raka jelita grubego. Obejmuje ona rektosigmoidoskopię oraz kolonoskopię. Kolonoskopia umożliwia uwidocznienie śluzówki całego jelita grubego, a rektosigmoidoskopia ze względu na zasięg badania może wykryć tylko jedną trzecią zmian i wymaga późniejszej oceny pozostałej bliższej części jelita[6]. Zawsze konieczne jest uwidocznienie całego jelita grubego, ponieważ istnieje ryzyko występowania guza synchronicznego[6][16].
Endoskopia umożliwia zobrazowanie i dokładne zlokalizowanie guza oraz pobranie materiału do badania histopatologicznego[286]. Ponadto umożliwia usunięcie większości zmian przednowotworowych[287]. Kolonoskopia wykazuje bardzo wysoką czułość i wysoką swoistość w wykrywaniu raka inwazyjnego oraz polipów, które mogą stanowić zmianę przednowotworową[286][288][289]. Skuteczność badania ogranicza złe przygotowanie chorego do badania i sytuacja niemożności zbadania całego jelita grubego[290].
Rak jelita grubego może być widoczny jako zmiana wypukła, płaska lub jako owrzodzenie. Endoskopowo zmiany klasyfikuje się według podziału Borrmanna i wyróżnia się postać polipowatą, grzybiastą (wrzodziejąco-polipowata), wrzodziejącą lub zmianę rozlaną. W celu ułatwienia rozpoznania bardzo wczesnych zmian bywają stosowane chemoendoskopia (barwienie błony śluzowej) oraz endoskopia powiększająca[290][291].
Przy niewielkich, płaskich zmianach pomocne bywa przedoperacyjne oznakowanie guza za pomocą założonego klipsa lub wykonania tatuażu[287].
Nierzadko pełna kolonoskopia nie może być wykonana z powodów technicznych lub stanu chorego. Powodem może być zwężenie nowotworowe lub zapalne jelita, silne uczucie dyskomfortu badanego, skurcz jelita, zrosty lub uchyłki[292].
Ultrasonografia endoskopowa (EUS) i ultrasonografia endorektalna (ERUS)
Ultrasonografia endoskopowa jest inwazyjną metodą diagnostyczną polegającą na umieszczeniu w przewodzie pokarmowym endoskopu wyposażonego w głowicę ultrasonograficzną. Ultrasonografia endorektalna jest podobną metodą i sonda jest umieszczana w obrębie odbytnicy[290]. Podstawowym celem badania jest ocena zaawansowania, a także rozpoznanie chorych wymagających chemioterapii neoadiuwantowej od chorych niewymagających tego leczenia[287].
Ultrasonografia endoskopowa w ocenie stopnia zaawansowania miejscowego raka jelita grubego (cecha T klasyfikacji TNM) charakteryzuje się bardzo wysoką czułością wynoszącą 80–96% i swoistością wynoszącą 75–98%[287][293][294][295] i wykazuje przewagę nad TK i MRI. W ocenie zajęcia węzłów chłonnych (cecha N klasyfikacji TNM) wykazuje około 70% czułość i 80% swoistość nie wykazując istotnej klinicznie przewagi nad TK i MRI[287][293][296]. Biopsja cienkoigłowa wykonana podczas EUS pomaga rozpoznać zajęcie węzłów chłonnych[287].
Endoskopia kapsułkowa
Endoskopia kapsułkowa jest metodą diagnostyczną, w której stosuje się kapsułkę zawierającą urządzenie rejestrujące (endoskop). W celu diagnostyki jelita grubego stosuje się inne urządzenia niż do diagnostyki jelita cienkiego. Kapsułka jest wyposażona w dwa aparaty i posiada zasilanie na około 10 godzin pracy. Kapsułkę endoskopową połyka się, w celu ograniczenia zużycia baterii urządzenie pozostaje w uśpieniu do około godziny, następnie podczas pasażu przez jelito grube wykonuje liczne zdjęcia śluzówki z częstością 2–4 zdjęć na sekundę. Ze względu na niepełne pole widzenia dwóch kamer część błony śluzowej pozostaje niezbadana. Ograniczeniem jest także niemożliwość wykonania dodatkowych obrazów z podejrzanego miejsca i niemożliwość wykonania zabiegów, w tym pobrania próbki do badania histopatologicznego[297][298]. Endoskopy kapsułkowe drugiej generacji w wykrywaniu polipów większych od 6 mm osiągają czułość wynoszącą do 86%[299][300][301].
Endoskopia kapsułkowa może być stosowana u chorych z niskim ryzykiem raka jelita grubego, bez objawów alarmujących i występowania raka jelita grubego w rodzinie. U chorych z wysokim ryzykiem rozpoznania raka, z objawami alarmującymi lub występowaniem raka jelita grubego w rodzinie zaleca się wykonanie kolonoskopii. Endoskopia kapsułkowa może być zastosowana u chorych, u których niemożliwe jest wykonanie pełnej kolonoskopii[302][298]. Endoskopia kapsułkowa może znaleźć zastosowanie jako badanie przesiewowe raka jelita grubego[299].
Wirtualna kolonoskopia TK i tomografia komputerowa
Wirtualna kolonoskopia jest badaniem radiologicznym, w którym na podstawie przekrojów uzyskanych w tomografii komputerowej tworzy się odwzorowanie powierzchni błony śluzowej. Zabieg wymaga dobrego przygotowania i wprowadzenia gazu (zwykle powietrza lub dwutlenku węgla) do jelita grubego w celu rozciągnięcia błony śluzowej[303].
Czułość badania jest uzależniona od wielkości zmiany. Dla polipów poniżej 6 mm czułość wynosi około 48%, dla polipów pomiędzy 6–9 mm czułość wynosi około 70%, a dla zmian większych niż 9 mm 85%. Swoistość dla polipów o wielkości poniżej 6 mm wynosi 92%, dla polipów o wielkości 6–9 mm 93% i 97% dla polipów powyżej 9 mm[303]. W metaanalizie kilku badań czułość wirtualnej kolonoskopii TK w wykrywaniu raka jelita grubego wynosiła 96% i była porównywalna z kolonoskopią[304][292].
Wirtualna kolonoskopia TK jest stosowana w przypadku niemożności wykonania pełnej kolonoskopii lub przeciwwskazań do badania. Metoda jest przydatna w ramach wstępnej diagnostyki u starszych chorych i chorych w złym stanie ogólnym z niepokojącymi objawami[292]. Podejrzenie raka jelita grubego za pomocą wirtualnej kolonoskopii wymaga wykonania optycznej kolonoskopii, która umożliwia pobranie materiału do badania histopatologicznego[6].
Metoda może znaleźć zastosowanie jako alternatywa do kolonoskopii w badaniach przesiewowych w kierunku raka jelita grubego. W Europie bywa wykorzystywana u chorych z niecałkowitym badaniem kolonoskopowym w ramach zastąpienia badania na krew utajoną w kale lub u chorych niezgadzających się na kolonoskopię[305][292].
Klasyczna wielorzędowa tomografia komputerowa jamy brzusznej, miednicy i klatki piersiowej jest wykorzystywana do oceny zaawansowania choroby nowotworowej[6][305][306].
Wirtualna kolonoskopia MRI i rezonans magnetyczny
Wirtualna kolonoskopia MRI bywa wykorzystywana do rozpoznawania raka jelita grubego u chorych, u których nie można wykonać pełnej kolonoskopii[303]. Metoda jest bardzo czuła w wykrywaniu polipów większych niż 10 mm i raka jelita grubego[307].
Rezonans magnetyczny jest stosowany do oceny zaawansowania choroby. Jest przydatny do oceny zaawansowania miejscowego guza (cecha T klasyfikacji TNM) oraz oceny obecności przerzutów w wątrobie (cecha M klasyfikacji TNM). Metoda jest szczególnie przydatna do oceny rozprzestrzeniania się guza poza światło narządu w obrębie odbytnicy, w tym oceny mezorektum (tkanki okołoodbytnicze) i powięzi mezorektum[308][309][310]. W ocenie zaawansowania miejscowego metoda nie wykazuje przewagi nad endorektalnym USG, które ma podobną skuteczność. Ze względu na wysokie koszty i ograniczoną dostępność MRI badania nie stosuje się rutynowo w ocenie stopnia zaawansowania miejscowego[305]. Rezonans magnetyczny wykazuje pewną przewagę nad tomografią komputerową w wykrywaniu przerzutów do wątroby[311][305].
Pozytonowa tomografia emisyjna
Pozytonowa tomografia emisyjna w połączeniu z tomografią komputerową (PET-TK) jest przydatną metodą w ocenie zaawansowania choroby, odpowiedzi na leczenie i rozpoznania nawrotu choroby[312][313]. Jest stosowana w ocenie obecności przerzutów pozawątrobowych, a wraz z podwyższonym stężeniem markera CEA wskazuje na nawrót[160]. Metoda jest oparta o wykrywanie zwiększonego wychwytu glukozy znakowanej fluorem (18F-FDG), co pozwala rozpoznać miejsca o zwiększonym poziomie metabolizmu, jakimi są ogniska nowotworu złośliwego[314].
Radiologiczne badanie dwukontrastowe
Badanie polega na podaniu zawiesiny siarczanu baru jako pozytywnego środka cieniującego oraz dwutlenku węgla jako negatywnego środka kontrastującego i wykonanie serii zdjęć rentgenowskich w różnych układach[315]. Czułość badania dla zmian mniejszych niż 5 mm wynosi 32%, a dla zmian 6–10 mm wynosi 50%[316]. Czułość badania w rozpoznawaniu raka esicy i odbytnicy wynosi około 80% i specyficzność wynosi około 98%[289].
W badaniu radiologicznym rak może się objawiać[317]:
- okrężnym zwężeniem przypominającym ogryzek jabłka,
- zmianami polipowatymi o grzybiastym kształcie,
- polipem z zagłębieniem u podstawy,
- polipem o nieregularnym zarysie,
- zmianą płaską z ubytkiem w kształcie płytki,
- zmianą z kraterowatym rozpadem i uniesionymi brzegami.
Markery nowotworowe
- CEA (antygen rakowo-płodowy)
CEA jest glikoproteiną stosowaną w diagnostyce wielu nowotworów złośliwych. Stężenie markera ulega podwyższeniu o 50% u chorych na raka jelita grubego, jednak zwykle następuje to w zaawansowanym stadium choroby[286]. Wysokie przedoperacyjne stężenie jest związane z większym zaawansowaniem choroby[318][319] i wyższym ryzykiem nawrotu[320]. Podwyższone stężenie CEA koreluje z gorszym rokowaniem[286][321]. Powtarzalne oznaczenia CEA wykazuje wysoką czułość w diagnostyce nawrotu choroby[321][322].
- CA 19-9
CA 19-9 jest glikoproteiną wykorzystywaną w diagnostyce przede wszystkim raka trzustki, jego podwyższone stężenie obserwuje się również w raku dróg żółciowych i innych nowotworach przewodu pokarmowego[286].
CA 19-9 wykorzystuje się w diagnostyce i monitorowaniu raka jelita grubego. Połączenie CEA i CA 19-9 dodatkowo zwiększa czułość badania[323][324][286][325]. CA 19-9 samodzielnie lub w połączeniu z CEA jest używany w monitorowaniu odpowiedzi na leczenie[326][327][328].
- CA 72-4
CA 72-4 jest glikoproteiną, którego podwyższone stężenie obserwuje się w różnych nowotworach złośliwych. Marker wykazuje bardzo wysoką swoistość sięgającą 100%, jednak jego przydatność ogranicza niewielką czułość wynosząca około 60%[329]. Skuteczność badania zwiększa jednoczesny pomiar CA 72-4 i CEA[286]. Wysokie stężenie CA 72-4 koreluje z wyższym zaawansowaniem nowotworu[330][331][330], wyższym ryzykiem nawrotu i gorszym rokowaniem[332].
- TPA
Tkankowy antygen polipeptydowy jest polipeptydem powstającym podczas fazy S i fazy G2 cyklu komórkowego. Jest uwalniany do krążenia w wyniku rozpadu komórek[333][286]. Zwiększone stężenie TPA jest stwierdzane w 60–80% przypadków raka jelita grubego[334][286]. Prawdopodobnie jego podwyższone stężenie jest niekorzystnym czynnikiem rokowniczym[334].
Biopsja

Zmiany rozpoznane podczas endoskopii lub badań obrazowych wymagają wykonania biopsji w celu potwierdzenia lub wykluczenia obecności nowotworu złośliwego. Pobrany bioptat jest następnie badany histopatologicznie. Celna biopsja może jednoznacznie potwierdzić raka, jednak pobrana próbka może nie dostarczać pełnej informacji o nowotworze[335].
Badanie histopatologiczne
Badanie histopatologiczne umożliwia ostateczne rozpoznanie choroby, które jest konieczne przed rozpoczęciem leczenia ogólnoustrojowego[336]. Badanie umożliwia określenie histologicznego typu guza, stopnia jego złośliwości histologicznej, histopatologiczną ocenę stopnia zaawansowania, w tym ocenę głębokości nacieku, zajęcia węzłów chłonnych, obecności przerzutów odległych oraz ocenę marginesów chirurgicznych[337].
Ocena zaawansowania i rokowania
Ocena zaawansowania choroby

Samo rozpoznanie choroby nowotworowej nie jest wystarczające do wyboru właściwego leczenia, do czego konieczna jest ocena zaawansowania choroby (ang. staging). Zaawansowanie choroby jest określane według klasyfikacji TNM, która składa się ze stopnia zaawansowania guza pierwotnego, stopnia zajęcia lokalnych węzłów chłonnych oraz obecności przerzutów odległych. Obecnie obowiązuje 7 klasyfikacja TNM według AJCC[336][338][339].
Ocena zaawansowania choroby początkowo jest przeprowadzana klinicznie na podstawie historii choroby, badania lekarskiego, kolonoskopii z biopsją oraz badań obrazowych. Następnie po chirurgicznym usunięciu guza preparat po operacji jest badany histopatologicznie, co jest podstawą do postawienia histologicznej oceny zaawansowania[340].
Kliniczna ocena guza pierwotnego (cecha T) jest przeprowadzana za pomocą ultrasonografii endoskopowej lub rezonansu magnetycznego[305]. Tomografia komputerowa, rezonans magnetyczny i ultrasonografia endoskopowa służą do oceny obecności przerzutów do pobliskich węzłów chłonnych (cecha N). W ocenie obecności przerzutów odległych wykorzystuje się tomografię komputerową, rezonans magnetyczny i pozytonową tomografię emisyjną[305]. Rezonans magnetyczny wykazuje pewną przewagę nad tomografią komputerową i PET w wykrywaniu przerzutów do wątroby[311]. W ocenie obecności przerzutów do płuc zaleca się RTG klatki piersiowej lub tomografię komputerową klatki piersiowej. Choć tomografia komputerowa wykazuje wyższą czułość w wykrywaniu przerzutów do płuc, to jednak ze względu na częste stwierdzanie zmian nienowotworowych w płucach, chorzy bez przerzutów wątrobie lub w węzłach chłonnych nie odnoszą korzyści z przedoperacyjnej tomografii komputerowej klatki piersiowej[341][305]. Wytyczne ESMO nie zalecają rutynowego wykonywania tomografii komputerowej klatki piersiowej[342].
Podstawowymi badaniami wykorzystywanymi do oceny zaawansowania choroby jest tomografia komputerowa jamy brzusznej i miednicy, zdjęcie rentgenowskie klatki piersiowej lub tomografia komputerowa klatki piersiowej, ewentualnie tomografia komputerowa klatki piersiowej[336]. Rezonans magnetyczny może być korzystniejszym badaniem u chorych z zaawansowanym nowotworem ze względu na lepszą czułość w wykrywaniu przerzutów do wątroby i otrzewnej[343]. PET-TK nie jest zalecany do rutynowej oceny zaawansowania klinicznego[342][344]. Przed operacją oznacza się stężenia CEA, brak normalizacji po miesiącu od zabiegu sugeruje nieskuteczność leczenia[342].
Czynniki rokownicze

Rokowanie chorego jest uzależnione od wielu czynników. Zależy ono od głębokości nacieku, położenia guza, zajęcia lokalnych węzłów chłonnych, obecności przerzutów odległych, stopnia zaawansowania klinicznego, stopnia złośliwości histopatologicznej, uzyskania ujemnych marginesów chirurgicznych i obecności nawrotu choroby[345].
Rokowanie chorego koreluje ze stopniem zaawansowania klinicznego guza ocenianego w klasyfikacji TNM. Kluczowa rokowniczo jest głębokość inwazji w głąb ściany jelita grubego[346]. Wraz z głębokością nacieku ściany narządu wzrasta ryzyko wznowy po leczeniu radykalnym, zajęcia węzłów chłonnych oraz obecności przerzutów odległych[346][346][347]. Sama wielkość guza nie koreluje z rokowaniem lub ma marginalny wpływ na rokowanie[346][348][349][350], choć wielkość nowotworu wpływa na miejscową kontrolę choroby[346]. Rozległa inwazja warstwy podśluzowej i obecność depozytów komórek nowotworowych poza guzem pogarszają rokowanie[347][351][346].
Do czynników rokowniczych zalicza się lokalizację guza. Nowotwory zlokalizowane w okrężnicy rokują lepiej od występujących w odbytnicy[177]. Gorsze rokowanie cechuje guzy o wrzodziejącym lub siedzącym typie makroskopowym w porównaniu do polipowatego typu wzrostu[352]. Egzofityczny wzór wzrostu jest korzystnym czynnikiem rokowniczym, wiąże się z mniejszym zaawansowaniem i mniejszym ryzykiem obecności przerzutów odległych. Niedrożność jelita oraz perforacja są niekorzystnymi czynnikami rokowniczymi[183]. Zajęcie lokalnych węzłów chłonnych jest bardzo ważnym, niekorzystnym czynnikiem rokowniczym, na przeżycie niekorzystnie wpływa sama obecność zajęcia węzłów chłonnych oraz rozległość ich zajęcia[346]. Nieuzyskanie marginesów chirurgicznych wolnych od nacieku wiąże się z wysokim ryzykiem nawrotu i gorszym rokowaniem[345]. Rokownicze znaczenie ma wzór naciekania nowotworu[353].
Prawdopodobnie podtyp histopatologiczny guza, z wyjątkiem tych z definicji o wysokim stopniu złośliwości, nie wpływa na rokowanie[349][354][355]. Stopień złośliwości histologicznej koreluje z rokowaniem[345]. Nowotwory słabo zróżnicowane (o wysokim stopniu złośliwości histologicznej) są związane z gorszym rokowaniem i słabszym przeżyciem chorych[170][347]. Gruczolakorak śluzowy i sygnetowaty są traktowane jak guzy o wyższym stopniu złośliwości i są skorelowane z gorszym przeżyciem[345][349]. Inwazja naczyń żylnych i limfatycznych pogarsza rokowanie, jest związana z wyższym ryzykiem nawrotu choroby oraz gorszym przeżyciem chorych[356][347]. Niezależnym niekorzystnym czynnikiem prognostycznym jest inwazja okołonerwowa, która znacząco zwiększa ryzyko wznowy i pogarsza przeżycie chorych[357][349].
Klasyfikacja TNM
Klasyfikacja TNM jest systemem oceny zaawansowania klinicznego nowotworu.
| Guz pierwotny – cecha T | |
| Tx | Nie można ocenić guza pierwotnego |
| T0 | Nie stwierdza się guza pierwotnego |
| Tis | Rak in situ |
| T1 | Guz nacieka warstwę podśluzową |
| T2 | Guz nacieka błonę mięśniową właściwą |
| T3 | Guz nacieka przez błonę mięśniową właściwą tkanki okołookrężnicze lub okołoodbytnicze |
| T4 | – |
| T4a | Guz powoduje perforację otrzewnej |
| T4b | Guz bezpośrednio nacieka lub przylega do innych narządów lub struktur |
| Zajęcie okolicznych węzłów chłonnych – cecha N | |
| Nx | Nie można ocenić okolicznych węzłów chłonnych |
| N0 | Nie stwierdza się przerzutów w regionalnych węzłach chłonnych |
| N1 | Obecne są przerzuty w 1–3 regionalnych węzłach chłonnych |
| N1a | Przerzut w 1 regionalnym węźle chłonnym |
| N1b | Przerzuty w 2–3 regionalnych węzłach chłonnych |
| N1c | Obecny depozyt lub depozyty komórek nowotworowych w warstwie podsurowiczej, krezce lub niepokrytych otrzewną tkankach okołookrężniczych lub okołoodbytniczych bez przerzutu lub przerzutów w węzłach chłonnych |
| N2 | Obecne są przerzuty w 4 lub więcej regionalnych węzłach chłonnych |
| N2a | Przerzuty w 4–6 regionalnych węzłach chłonnych |
| N2b | Przerzuty w 7 lub więcej regionalnych węzłach chłonnych |
| Przerzuty odległe – cecha M | |
| Mx | Nie można określić obecności przerzutów odległych |
| M0 | Nie stwierdza się przerzutów odległych |
| M1 | Obecne przerzuty odległe |
| M1a | Przerzuty ograniczone do jednego narządu lub miejsca (np. wątroba, płuco, jajnik, pozaregionalny węzeł chłonny) |
| M1b | Przerzuty w więcej niż jednym narządzie lub miejscu lub w otrzewnej |
| Stopień zaawansowania | Cecha T | Cecha N | Cecha M |
| 0 | Tis | N0 | M0 |
| IA | T1 | N0 | M0 |
| T2 | N0 | M0 | |
| IIA | T3 | N0 | M0 |
| IIB | T4a | N0 | M0 |
| IIC | T4b | N0 | M0 |
| IIIA | T1 | N1 | M0 |
| T2 | N1 | M0 | |
| T1 | N2a | M0 | |
| IIIB | T3 | N1 | M0 |
| T4a | N1 | M0 | |
| T2 | N2a | M0 | |
| T3 | N2a | M0 | |
| T1 | N2b | M0 | |
| T2 | N2b | M0 | |
| IIIC | T4a | N2a | M0 |
| T3 | N2b | M0 | |
| T4a | N2b | M0 | |
| T4b | N1 | M0 | |
| T4b | N2 | M0 | |
| IVa | dowolne T | dowolne N | M1a |
| IVb | dowolne T | dowolne N | M1b |
Inne klasyfikacje
Obok klasyfikacji TNM, która jest zalecaną klasyfikacją do oceny zaawansowania choroby, bywają używane starsze klasyfikacje Dukesa i Astler-Collera[360].
| Stopień | Cechy |
| A | Nowotwór nie przekracza ściany jelita |
| B | Nowotwór przekracza ścianę jelita do surowicówki lub tkanki tłuszczowej okołoodbytniczej |
| C | Przerzuty w okolicznych węzłach chłonnych |
| D | Przerzuty odległe |
| Stopień | Cechy |
| A | Rak ograniczony do błony śluzowej |
| B1 | Naciek nie przekracza ściany mięśniowej |
| B2 | Naciek przekracza ścianę mięśniową, obejmuje tkanki okołookrężnicze lub otrzewną |
| C1 | Stopień B1 i zajęcie regionalnych węzłów chłonnych |
| C2 | Stopień B2 i zajęcie regionalnych węzłów chłonnych |
| D | Obecne przerzuty odległe |
Leczenie
Strategia leczenia raka jelita grubego zależy od stopnia zaawansowania choroby, stanu ogólnej sprawności chorego oraz współistniejących istotnych chorób. Całkowite wyleczenie raka jelita grubego jest możliwe wyłącznie u chorych poddanych radykalnemu zabiegowi chirurgicznemu, w pozostałych przypadkach leczenie jest ukierunkowane przede wszystkim na maksymalne wydłużenie przeżycia chorego. Zabieg chirurgiczny polega na szerokim wycięciu guza z zachowaniem odpowiednich marginesów zdrowych tkanek[10]. Rodzaj zabiegu zależy od położenia guza, a w przypadku guzów położonych w odbytnicy także od możliwości jego usunięcia z zachowaniem funkcji zwieraczy odbytu. W bardzo wczesnych stadiach choroby rak może być leczony endoskopowo[363][364]. W przypadku raka okrężnicy w zależności od położenia guza wykonuje się częściową prawostronną lub lewostronną hemikolektomię albo odcinkową resekcję okrężnicy[365]. W przypadku raka odbytnicy wykonuje się przednią resekcję odbytnicy lub brzuszno-kroczową amputację odbytnicy. Zachowanie zwieraczy warunkuje bliskość nacieku raka w stosunku do zwieraczy, konieczność zachowania odpowiedniego marginesu wolnego od nacieku nowotworowego oraz techniczna możliwość utworzenia zespolenia[366]. W raku odbytnicy standardowym elementem każdego zabiegu jest całkowite wycięcie mezorektum[367].
Leczenie okołooperacyjne różni się w raku okrężnicy i w raku odbytnicy. Celem leczenia okołooperacyjnego jest zmniejszenie ryzyka nawrotu, wydłużenie przeżycia chorych i w przypadku raka odbytnicy ułatwienie lub umożliwienie wykonania radykalnego zabiegu z zachowaniem zwieraczy. Leczenie operacyjne raka odbytnicy w zaawansowanym stadium choroby, obejmujące wysokie zaawansowanie miejscowe choroby (guzy T3–T4), zajęcie lokalnych węzłów chłonnych lub lokalnie nieoperacyjny guz, wymaga zastosowania leczenia neoadiuwantowego lub ewentualnie jest uzupełniane leczeniem adiuwantowym, gdy leczenie neoadiuwantowe nie było stosowane przed operacją. Leczenie neoadiuwantowe w raku odbytnicy polega na chemioradioterapii z kapecytabiną lub 5-fluorouracylem albo 5-fluorouracylem z leukoworyną, a następnie chemioterapii z użyciem programu FOLFOX, kapecytabiny z lub bez oksaliplatyny albo 5-fluorouracylu z lub bez leukoworyny. Chemioradioterapia może być poprzedzona chemioterapią[368]. U chorych w pośrednim zaawansowaniu raka odbytnicy równoważnym leczeniem jest krótkotrwała radioterapia bez chemioterapii[369][370]. Leczenie raka okrężnicy w stadium III zaawansowania klinicznego według TNM jest uzupełniane o sześciomiesięczną chemioterapię adiuwantową za pomocą 5-fluorouracylu, kapecytabiny, programu FOLFOX, CapeOx albo FLOX[371]. W niektórych przypadkach konieczne jest również włączenie leczenia adiuwantowego w stadium II zaawansowania klinicznego[10].
Leczenie choroby z przerzutami jest oparte o chemioterapię, jej charakter zależy od założonych celów terapeutycznych i stanu chorego[13]. U niewielkiej części chorych możliwe jest wykonanie chirurgicznego wycięcia pojedynczych przerzutów do wątroby lub płuc, a pewna część chorych może odnieść korzyść z intensywnej chemioterapii mającej na celu zmniejszenie wielkości przerzutów i techniczne umożliwienie operacji. U większości chorych operacja nie jest możliwa i intensywna wielolekowa chemioterapia ma na celu przede wszystkim jak największe wydłużenie przeżycia chorych[372]. W ramach intensywnej chemioterapii stosuje się wielolekowe programy lecznicze oparte o fluoropirymidynę (5-fluorouracyl, kapecytabina) w połączeniu z oksaliplatyną lub irynotekanem tworząc schematy FOLFOX, CapeOX (XELOX), FOLFIRI i FOLFOXIRI, które mogą być kojarzone z bewacyzumabem lub inhibitorami EGFR (cetuksymab, panitumumab)[373][15]. U chorych w złym stanie klinicznym podstawowym celem jest złagodzenie objawów choroby i hamowanie progresji choroby, w tym celu stosuje się leczenie oparte o pojedyncze aktywne w raku jelita grubego leki przeciwnowotworowe[372].
Leczenie chirurgiczne w chorobie ograniczonej
Leczenie endoskopowe
Większość przypadków raka jelita grubego jest stwierdzana podczas kolonoskopii wykonanej przesiewowo lub z powodu niewyjaśnionych dolegliwości. Choć badanie pozwala klinicznie postawić podejrzenie nowotworu złośliwego, to jednak nie ma możliwości wykluczenia go bez wykonania biopsji i wykonania badania histopatologicznego. W związku z tym wszystkie stwierdzone podczas endoskopii podejrzane zmiany są w miarę możliwości usuwane w całości, a następnie badane histopatologicznie[374][375]. Rodzaj zabiegu endoskopowego zależy od wielkości i typu wzrostu polipa, a w zależności od histopatologicznych cech rokowniczych ewentualne dalsze postępowanie[376].
Małe polipy o wielkości do 5–8 mm są usuwane w całości za pomocą kleszczyków biopsyjnych. Większe, uszypułowane polipy są usuwane za pomocą pętli diatermicznej lub elektrokoagulacji. Kluczowe jest oznaczenie na preparacie miejsca wycięcia polipa, ponieważ determinuje to ocenę marginesu chirurgicznego i wybór dalszego postępowania. Duże zmiany o siedzącym typie wzrostu (o szerokiej podstawie) nie są usuwane podczas endoskopii, ale pobiera się z nich wycinek do badania histopatologicznego. w przypadku polipów, które są zbyt duże aby je usunąć podczas zabiegu endoskopowego i łagodnym charakterze pobranej biopsji w badaniu histopatologicznym, przeprowadza się odcinkową resekcję jelita grubego z marginesem 5 cm[376].
Rak o charakterze uszypułowanego lub siedzącego polipa z korzystnymi czynnikami ryzyka („rak w polipie”, „złośliwy polip”) po endoskopowym całkowitym usunięciu nie wymaga dalszego leczenia poza leczeniem endoskopowym. Do korzystnych czynników ryzyka zalicza się osiągnięcie ujemnych marginesów chirurgicznych, 1 i 2 stopień złośliwości histologicznej oraz bez inwazji struktur naczyniowych[363]. Jednak w przypadku polipa siedzącego po leczeniu endoskopowym, ze względu na większe prawdopodobieństwo nawrotu i zajęcia węzłów chłonnych, pomimo ujemnych marginesów i korzystnych czynników ryzyka histopatologicznego standardową metodą leczenia jest resekcja jelita grubego[375][377].
Jeśli wiarygodna ocena marginesów jest niemożliwa lub badanie histopatologiczne wykaże niekorzystne cechy rokownicze, to konieczna jest resekcja jelita grubego z wycięciem en block węzłów chłonnych[363]. Do niekorzystnych cech histopatologicznych wymagających bardziej rozległego leczenia zalicza się brak wolnych od nacieku marginesów chirurgicznych wycięcia, 3 i 4 stopień złośliwości histologicznej i inwazja struktur naczyniowych[363][378]. Za ujemny margines przyjmuje się brak nacieku nowotworowego 1–2 mm linii cięcia[363]. Stwierdzenie marginesów wolnych od nacieku nowotworowego, braku naciekania struktur naczyniowych i 1–2 stopnia złośliwości histologicznej nie wyklucza możliwości wznowy po leczeniu endoskopowym[375].
Klasyczna operacja




U chorych z chorobą operacyjną bez przerzutów odległych wykonuje się zabieg operacyjny polegający na anatomicznej resekcji jelita grubego z guzem w jednym bloku tkankowym (en block) z regionalnymi węzłami chłonnymi. Zakres zabiegu jest uzależniony od położenia guza, anatomicznej struktury unaczynienia tętniczego i limfatycznego[379][376].
W przypadku guza położonego w kątnicy, wstępnicy i zgięcia wątrobowego wykonuje się prawostronną hemikolektomię. Zabieg polega na usunięciu 10–15 cm końcowego odcinka jelita krętego, całej kątnicy, wstępnicy i bliższego odcinka poprzecznicy. Ciągłość przewodu pokarmowego odtwarza się za pomocą zespolenia jelita krętego z pozostawioną częścią poprzecznicy. Ze względu na specyfikę unaczynienia i konieczność usunięcia określonego dorzecza węzłów chłonnych nie przeprowadza się mniej rozległych operacji. Zamykane są niektóre odgałęzienia tętnicy krezkowej górnej: tętnica krętniczo-kątnicze, tętnica krętnicza prawa i prawa gałąź tętnicy okrężniczej środkowej. Jeśli występuje konieczność podwiązania lewej gałęzi tętnicy okrężniczej środkowej to konieczna jest kontrola unaczynienia pozostawianej części poprzecznicy i ewentualne poszerzenie resekcji w przypadku braku odpowiedniego ukrwienia. Guzy położone w początkowym odcinku poprzecznicy wymagają usunięcia całej tętnicy okrężniczej środkowej, prawej tętnicy okrężniczej i tętnicy krętniczo-kątniczej[380][381][382][383].
Guzy położone w poprzecznicy wymagają różnego sposobu leczenia chirurgicznego w zależności od położenia[384]. W przypadku guzów położonych w okolicy zgięcia wątrobowego oraz środkowej części okrężnicy wykonuje się rozszerzoną prawostronną hemikolektomię poszerzoną o podwiązanie prawego i lewego odgałęzienia tętnicy okrężniczej środkowej[385]. W niektórych przypadkach, gdy jelito grube jest dobrze ruchome, guzy środkowej części poprzecznicy mogą być leczone odcinkową resekcją poprzecznicy[382][381]. Jednak taka operacja jest rzadko wykonywana ze względu na niewystarczającą szerokość resekcji oraz ryzyko powstania wewnętrznej przepukliny[386]. Prawostronna hemikolektomia zapewnia lepsze ukrwienie zespolenia, co przekłada się na mniejsze ryzyko jego nieszczelności[384]. Guzy położone w dalszym odcinku poprzecznicy i zgięcia śledzionowego wymagają lewostronnej hemikolektomii[386][381].
W przypadku guzów zlokalizowanych w zstępnicy wykonuje się lewostronną hemikolektomię. Zabieg polega na usunięciu zgięcia śledzionowego i wzstępnicy, z podwiązaniem tętnicy okrężniczej lewej (odgałęzienie tętnicy krezkowej dolnej) i wykonaniu zespolenia pomiędzy poprzecznicą a esicą[387][388][382].
Guzy esicy mogą być leczone resekcją esicy z zachowaniem 5 cm marginesów chirurgicznych po mobilizacji poprzecznicy i zgięcia śledzionowego i podwiązaniu tętnicy krezkowej dolnej poniżej oddzielenia się tętnicy okreżniczej lewej[389][390][387][382].
Rak odbytnicy może być leczony przez przezodbytowe wycięcie guza, przednią resekcję odbytnicy albo brzuszno-kroczową amputacje odbytnicy. Zawsze dąży się do zachowania zwieraczy, choć nie zawsze jest to możliwe[391]. Preferowaną operacją jest przednia resekcja odbytnicy, która pozwala zachować funkcję zwieraczy odbytu, jednak nie jest ona możliwa do wykonania u wszystkich chorych[392].
Przezodbytnicze wycięcie guza może być przeprowadzone w wybranych przypadkach guza o niewielkim zaawansowaniu miejscowym bez cech zajęcia węzłów chłonnych (guzy T1N0M0). Nowotwór musi się znajdować do 8 cm od brzegu odbytu, nie przekraczać 3 cm ani nie może zajmować więcej niż 30% obwodu odbytnicy[393]. Nowotwór musi być dobrze lub umiarkowanie zróżnicowany i w biopsji nie mogą występować cechy inwazji żylnej lub limfatycznej[394][393][395]. Jeśli w badaniu histopatologicznym stwierdza się niekorzystne cechy guza zwiększające ryzyko nawrotu, to wykonuje się radykalną resekcję odbytnicy[396][397]. Wycięcie musi zachować odpowiednio szeroki margines zdrowych tkanek i musi być głębokie sięgając do okołoodbytniczej tkanki tłuszczowej[398]. Margines powinien wynosić przynajmniej 1 cm[396]. Ograniczeniem metody jest obecność mikroprzerzutów do okolicznych węzłów chłonnych[399]. Rak odbytnicy w stadium T1 charakteryzuje się większą częstością zajęcia lokalnych węzłów chłonnych (13–25%) niż rak w okrężnicy (3–8%)[400][401][396]. Z tego względu przed zabiegiem konieczne jest wykonanie ultrasonografii endorektalnej (ERUS)[396]. Wycięcie przezodbytnicze jest metodą mniej inwazyjną i obciążającą dla chorego, jednak odsetek nawrotów tej metody leczenia jest większy niż w przypadku bardziej rozległej operacji radykalnej[399][402][403][398] oraz oferuje nieznacznie gorsze przeżycie całkowite leczonych niż chorych leczonych operacją radykalną[404][399]. Przezodbytnicza mikrochirurgia endoskopowa (TEN) może ułatwiać wycięcie przez odbyt niewielkich guzów[393].
W guzach o wyższym zaawansowaniu (T2, T3) leczenie chirurgiczne polega na przedniej resekcji odbytnicy lub brzuszno-kroczowej amputacji odbytnicy[391]. Guzy położone powyżej załamka otrzewnej są leczone na podobnych zasadach jak rak w esicy[398]. Guzy położone 1–2 cm od linii zębatej mogą być wycięte z odtworzeniem ciągłości przewodu pokarmowego bez wytwarzania stałej kolostomii[367]. Przed operacją u niektórych chorych stosuje się neoadiuwantową radiochemioterapię w celu zwiększenia szansy na wykonanie zabiegu oszczędzającego zwieracze[367].
Przednia resekcja odbytnicy jest stosowana w zmianach dotyczących górnej i środkowej części odbytnicy. Operacja polega na mobilizacji i przecięciu odbytnicy, a następnie odtworzeniu ciągłości przewodu pokarmowego za pomocą zespolenia bok-do-końca[405][406][407]. Podczas resekcji nisko położonego guza w celu zachowania zwieraczy możliwe jest wykonanie węższego marginesu zdrowych tkanek. Minimalny margines cięcia powinien wynosić przynajmniej 1–2 cm powyżej zwieraczy[398][391]. W celu poprawy wielkości marginesu chirurgicznego bywają wykonywane operacje z częściową resekcją zwieraczy z zachowaniem ich funkcji[408], jednak nie ma dużych badań potwierdzających skuteczność takiego postępowania[367].
Brzuszno-kroczowa (krzyżowa) amputacja odbytnicy (operacja Milesa) jest wykonywana w przypadku nacieku zwieraczy odbytu, mięśnia dźwigacza odbytu lub konieczny zakres resekcji spowoduje utratę funkcji zwieraczy odbytu lub nietrzymanie moczu. Brzuszno-kroczowa amputacja odbytnicy polega na wycięciu w jednym bloku tkankowym (en block) części esicy, odbytnicę, odbyt i mezorektum z wyłonieniem stałej kolostomii[392].
Kilka badań wskazuje, że operacja brzuszno-kroczowa jest związana z gorszą lokalną kontrolą choroby oraz gorszym przeżyciem całkowitym leczonych[409][410][392].
Zarówno w przedniej resekcji odbytnicy, jak i w brzuszno-kroczowej amputacji odbytnicy wykonuje się całkowite wycięcie mezorektum (TME)[392]. Jest to standardowa procedura w przypadku guzów położonych w dolnej i środkowej części odbytnicy. W przypadku guzów górnej części odbytnicy zaleca się wycięcie mezorektum do 5 cm od brzegu guza[391][411]. Zabieg polega na wypreparowaniu mezorektum wraz z powięzią mezorektum[412]. Całkowite wycięcie mezorektum zmniejsza ryzyko nawrotu miejscowego i wydłuża przeżycie całkowite leczonych[413][414]. Niecałkowite wycięcie mezorektum wiąże się z wyższym ryzykiem wznowy[415]. Ponadto w raku w dalszej części odbytnicy bez naciekania zwieraczy całkowite wycięcie mezorektum sprzyja zachowaniu funkcji zwieraczy odbytu. Operacja ułatwia zaoszczędzenie niektórych okolicznych nerwów, co przekłada się na zachowanie prawidłowej funkcji pęcherza moczowego oraz funkcji seksualnych[416][417]. W leczeniu raka odbytnicy nie zaleca się dodatkowego poszerzania usunięcia węzłów chłonnych (limfadenektomii), poza usunięciem węzłów podejrzanych klinicznie o obecność przerzutów węzłowych[399]. Podobnie w raku okrężnicy wycina się również wszystkie podejrzane węzły chłonne położone poza obszarem resekcji[379].
Operacja laparoskopowa

_1.jpg)
Operacja laparoskopowa jest opcją chirurgicznego leczenia raka okrężnicy. Operacje wykonywane metodą laparoskopową pozwalają osiągać podobne wyniki leczenia raka okrężnicy w porównaniu do metody otwartej[418][419][420][421][422][423]. Operacja laparoskopowa wiąże się z krótszym okresem pooperacyjnym i krótszym pobytem w szpitalu[424][421][379]. Metoda nie jest zalecana w przypadku guzów powodujących niedrożność, perforację lub naciekających sąsiednie struktury[425].
W przypadku raka odbytnicy operacja laparoskopowa prawdopodobnie oferuje podobne wyniki długoterminowe jak operacja metodą klasyczną[426][427][428][429].
Leczenie okołooperacyjne choroby potencjalnie operacyjnej bez przerzutów
W leczeniu raka jelita grubego stosuje się leczenie neoadiuwantowe poprzedzające leczenie operacyjne lub leczenie adiuwantowe, które podaje się po zabiegu chirurgicznym w celu poprawy skuteczności zabiegu. W związku z odmiennościami przebiegu klinicznego i specyfiką leczenia raka końcowego odcinka jelita grubego dla raka w okrężnicy i raka w odbytnicy przyjęto odmienne strategie leczenia okołooperacyjnego.
Rak okrężnicy
Leczenie adiuwantowe jest leczeniem stosowanym po przeprowadzonym radykalnym zabiegu chirurgicznym w celu zmniejszeniu ryzyka wznowy miejscowej lub pojawienia się przerzutów odległych.
U chorych z rakiem jelita grubego zasadność zastosowania leczenia adiuwantowego zależy od zaawansowania choroby[430]:
- W chorobie w stadium I klinicznym nie stosuje się leczenia adiuwantowego.
- W chorobie w stadium II klinicznym o niskim ryzyku może być zastosowane leczenie adiuwantowe za pomocą kapecytabiny lub 5-fluorouracylu albo chorych wyłącznie się obserwuje.
- W chorobie w stadium II klinicznym o wysokim ryzyku – obejmującym guz w stopniu zaawansowania miejscowego T4, o wysokiej złośliwości histologicznej (z wyłączeniem nowotworów z niestabilnością mikrosatelitarną), naciekiem marginesów chirurgicznych, inwazją naczyń krwionośnych i limfatycznych, naciekiem okołonerwowym, niedrożnością jelit, perforacją jelita lub niewystarczającą oceną węzłów chłonnych (poniżej 12 węzłów) – stosuje się leczenie adiuwantowe za pomocą 5-fluorouracylu lub kapecytabiny albo programu FOLFOX lub CapeOx albo FLOX lub wyłącznie się obserwuje.
- W chorobie w stadium III klinicznym u wszystkich chorych zaleca się leczenie adiuwantowe za pomocą programu FOLFOX lub CapeOx albo FLOX albo 5-fluorouracylu lub kapecytabiny w monoterapii.
Kilka badań na chorych w II i III stadium klinicznym wykazało skuteczność leczenia adiuwantowego u chorych w III stadium klinicznym[431]. Korzyści z leczenia adiuwantowego u chorych w II stadium klinicznym są znacznie mniejsze. Chorzy z przeciętnymi czynnikami ryzyka w II stadium klinicznym mają dobre rokowanie, dlatego nie odnoszą dużych korzyści z leczenia uzupełniającego. Z kolei chorzy z większym ryzykiem nawrotu odnoszą więcej korzyści z leczenia adiuwantowego[432]. Kilka badań wskazuje na korzyści z leczenia adiuwantowego w II stadium klinicznym[433][434][435]. Wyniki sugerują, że korzyści mogą być większe dla chorych z większym ryzykiem zajęcia węzłów chłonnych[433][432]. Z kolei w innym badaniu nie wykazano korzyści z leczenia adiuwantowego również u chorych z czynnikami niekorzystnego rokowania, choć ograniczeniem pracy był starszy wiek leczonych[436][432]. Wykazano, że opóźnienie chemioterapii adiuwantowej o 4 tygodnie powoduje zmniejszenie przeżycia całkowitego chorych[437].
U chorych w stadium II klinicznym zaleca się ocenę niestabilności mikrosatelitarnej (MSI), która jest czynnikiem korzystnego rokowania. Pozwala to na identyfikację chorych nie korzystających z leczenia adiuwantowego za pomocą 5-fluorouracylu. Ocenę niestabilności mikrosatelitarnej zaleca się również u wszystkich chorych ≤70. roku życia oraz u chorych z zespołem Lyncha[438].
Program FOLFOX6 jest preferowany nad programem FOLFOX4 w leczeniu adiuwantowym i choroby z przerzutami[439]. W badaniu klinicznym na 2246 chorych w III stadium klinicznym porównano skuteczność leczenia adiuwantowego za pomocą programu FOLFOX (5-fluorouracyl, leukoworyna i oksaliplatyna) z połączeniem 5-fluorouracylu z leukoworyną (folinian wapnia). Wykazano, że program FOLFOX wywoływał dłuższe 5-letnie przeżycie wolne od choroby (DFS) w porównaniu do 5-fluorouracylu z folinianem wapnia wynoszące odpowiednio 66,4% i 58,9%, a także dłuższe 6-letnie przeżycie całkowite (OS) leczonych za pomocą programu FOLFOX w porównaniu do 5-fluorouracylu wynoszące odpowiednio 72,9% i 68,7%[440]. Analiza pięciu badań na łącznie 4060 chorych poniżej 75. roku życia w III stadium klinicznym raka okrężnicy wykazała, że dodanie oksaliplatyny do 5-fluorouracylu poprawia przeżycie całkowite chorych[441]. Również inna analiza danych potwierdza pozytywny wpływ na przeżycie dodania oksaliplatyny do 5-fluorouracylu również u chorych po 75. roku życia[442][439].
Program FLOX również jest złożony z 5-fluorouracylu, leukoworyny i oksaliplatyny, jednak różni się od FOLFOX odmienną wielkością i rozkładem dawek. Skuteczność programu FLOX porównano z połączeniem 5-fluorouracylu i leukoworyny (FULV). FLOX wykazał wyższy odsetek 4-letnich przeżyć wolnych od choroby (DFS) wynoszący 73,2% w porównaniu do 67,0% dla FULV[443]. Po 7-letniej obserwacji program FLOX dalej wykazywał korzyści w wydłużeniu przeżycia wolnego od choroby, jednak nie obserwowano korzyści w wydłużeniu przeżycia całkowitego (OS)[444][445].
Programy FOLFOX i FLOX są podobne pod względem skuteczności, jednak różnią się profilem toksyczności i drogą podania. Program FLOX znacznie częściej wywołuje ciężką biegunkę niż program FOLFOX. Z kolei program FLOX nie wymaga podania do cewnika centralnego[446].
Kapecytabina jest doustnym prolekiem metabolizowanym do 5-fluorouracylu. W leczeniu adiuwantowym III stadium zaawansowania raka jelita grubego wywołuje nie gorsze przeżycie wolne od choroby (DFS) i przeżycie całkowite (OS) jak 5-fluorouracyl[447][448][445]. Kapecytabina może być łączona z oksaliplatyną (program CapeOX/XELOX). Połączenie wywołuje dłuższe 3-letnie przeżycie wolne od choroby (DFS) niż 5-fluorouracyl w połączeniu z leukoworyną[449][445].
U wybranych chorych z guzem o zaawansowaniu T4 według TNM stosuje się adiuwantową chemioradioterapię z użyciem 5-fluorouracylu. Może być zastosowana przedoperacyjna (neoadiuwantowa) chemioradioterapia w celu uzyskania operacyjności guza[450].
Rak odbytnicy
Neoadiuwantowe lub adiuwantowe leczenie raka odbytnicy jest przeprowadzane w stadium II i III zaawansowania klinicznego ze względu na wysokie ryzyko nawrotu choroby. Ryzyko nawrotu jest związane z trudnościami technicznymi uzyskania wystarczająco szerokiego marginesu chirurgicznego, braku przydanki odbytnicy i bliskości odbytnicy do innych narządów miednicy. Leczenie okołooperacyjne w raku odbytnicy polega na chemioradioterapii opartej na fluoropirymidynach i napromieniowaniu miednicy i następnie wykonaniu zabiegu radykalnego uzupełnionego chemioterapią[451]. Postępowanie okołooperacyjne zależy od rozpoznanego stopnia zaawansowania choroby[452]. Zaleca się by czas leczenia okołooperacyjnego wynosił około 6 miesięcy[453]. Zabieg u chorych w stadium T3 N0 M0 lub zajęcia węzłów chłonnych (cecha N1–N2) bez poprzedzającego leczenia neoadiuwantowego powinien być zarezerwowany wyłącznie dla chorych z przeciwwskazaniami do chemioradioterapii. Wówczas po radykalnym onkologicznie zabiegu po histopatologicznej ocenie stadium zaawansowania T1–T2 N0 M0 chorego wyłącznie się obserwuje, z kolei u chorych w stadium T3 N0 M0 lub chorych T1–T3 N0 M0 rozważa się zastosowanie leczenia adiuwantowego[453]
Rak odbytnicy oceniony przedoperacyjnie jako guz o zaawansowaniu miejscowym T1 lub T2[454][453]:
- w przypadku klinicznego zaawansowania choroby ocenionego T1–T2 N0 M0 po radykalnym zabiegu chirurgicznym nie jest konieczne leczenie okołoooperacyjne,
- w przypadku zaawansowania choroby ocenionego przed operacją jako T1–T2 N0 M0, ale wyższego patomorfologicznego zaawansowania po operacji ocenionego na T3 N0 M0 lub T1–T4 N1–N2 konieczne jest zastosowanie sześciomiesięcznego leczenia adiuwantowego na które składa się kolejno:
- chemioterapia za pomocą programu FOLFOX[a] albo kapecytabina z lub bez oksaliplatyny (CapeOX)[a] albo 5-fluorouracyl z lub bez leukoworyny,
- chemioradioterapia z kapecytabiną[a] albo chemioradioterapia z 5-fluorouracylem[a] lub chemioradioterapia z 5-fluorouracylem z leukoworyną,
- chemioterapia za pomocą programu FOLFOX[a] albo kapecytabiny z lub bez oksaliplatyny (CapeOX)[a] albo 5-fluorouracyl z lub bez leukoworyny,
- albo
U chorych z guzem o zaawansowaniu T3 N0 M0 z ujemnymi marginesami chirurgicznymi korzyści z radioterapii nie są duże i wystarczającym leczeniem może u nich być wyłącznie chemioterapia[453][455][455]. Jednak ze względu na częste niedoszacowanie zajęcia węzłów chłonnych mimo wykonania ERUS zaleca się rutynowe stosowanie radioterapii w stadium T3 N0[451].
W przypadku zaawansowanej choroby zabieg chirurgiczny może być wykonany bez poprzedzającego leczenia neoadiuwantowego i wówczas terapię uzupełnia się leczeniem adiuwantowym albo zabieg chirurgiczny jest poprzedzony leczeniem neoadiuwantowym, a następnie po zabiegu może być przeprowadzone leczenie adiuwantowe[456][453].
W przypadku choroby ocenionej przedoperacyjnie jako nowotwór zaawansowany (T3–T4), z zajęciem węzłów chłonnych (N1–N2) lub lokalnie nieoperacyjny[456][453]:
- w przypadku choroby lokalnie zaawansowanej T3–T4, lokalnie nieoperacyjnej lub zajęciem węzłów chłonnych (N1–N2) stosuje się leczenie okołooperacyjne w kolejności:
- chemioterapia za pomocą programu FOLFOX[a] albo kapecytabiny z lub bez oksaliplatyny (CapeOX)[a] albo 5-fluorouracylu z lub bez leukoworyny,
- chemioradioterapia z kapecytabiną[a] albo chemioradioterapia z 5-fluorouracylem[a] lub chemioradioterapia z 5-fluorouracylem z leukoworyną,
- zabieg operacyjny,
- albo
- chemioradioterapia z kapecytabiną[a] albo chemioradioterapia z 5-fluorouracylem[a] lub chemioradioterapia z 5-fluorouracylem z leukoworyną,
- zabieg operacyjny,
- chemioterapia adiuwantowa za pomocą programu FOLFOX[a] albo kapecytabiny z lub bez oksaliplatyny (CapeOX)[a] albo 5-fluorouracylu z lub bez leukoworyny.
- w przypadku przeciwwskazań do chemioradioterapii w przypadku zaawansowania choroby T3 N0 lub T1–T4 i N1–N2 lub lokalnie nieoperacyjnej po leczeniu chirurgicznym[457]:
- histopatologicznie rozpoznano stadium zaawansowania pT1–T2 N0 M0 i chorego wyłącznie obserwuje się,
- histopatologicznie rozpoznano stadium zaawansowania pT3–T4 N0 M0 lub pT1–T4 N1–N2 M0 i w dalszym postępowaniu rozważa się leczenie w kolejności:
- chemioterapia za pomocą programu FOLFOX[a] albo kapecytabiny z lub bez oksaliplatyny (CapeOX)[a] albo 5-fluorouracylu z lub bez leukoworyny,
- chemioradioterapia z kapecytabiną[a] albo chemioradioterapia z 5-fluorouracylem[a] lub chemioradioterapia z 5-fluorouracylem z leukoworyną,
- chemioterapia za pomocą programu FOLFOX[a] albo kapecytabiny z lub bez oksaliplatyny (CapeOX)[a] albo 5-fluorouracylu z lub bez leukoworyny,
- albo

Radioterapia lub radiochemioterapia jest kluczowym elementem leczenia okołooperacyjnego[12]. Radioterapia lub radiochemioterapia po zabiegu chirurgicznym jest następnie uzupełniana o chemioterapię adiuwantową[458]. Wskazania do radiochemioterapii obejmują dwie grupy chorych: chorych ze znacznym zaawansowaniem nowotworu i chorobą nieoperacyjną oraz chorych o pośrednim zaawansowaniu obejmującym chorobę o zaawansowaniu miejscowym T3 lub zajęciu lokalnych węzłów chłonnych (cecha N+)[369]. W chorobie o znacznym zaawansowaniu miejscowym stosuje się chemioradioterapię. W chorobie o pośrednim stopniu zaawansowaniu (T3, N+) może być stosowana radiochemioterapia z dawką około 50 Gy lub tylko krótkotrwała radioterapia z pięcioma dawkami po 5 Gy bez chemioterapii[369][370]. U starannie wybranych chorych w specjalistycznych ośrodkach może być wykonany zabieg chirurgiczny bez poprzedzającego leczenia neoadiuwantowego[459].
W zalecaniach NCCN zaleca się stosowanie przedoperacyjnej (neoadiuwantowej) chemioradioterapii u chorych w stadium II i III zaawansowania klinicznego. Pooperacyjna (adiuwantowa) radiochemioterapia jest stosowana u chorych z rakiem odbytnicy początkowo ocenionych jako stadium I, ale po operacji i badaniu histopatologicznym zmienioną kwalifikacją na stadium II lub III[460]. Neoadiuwantowa chemioradioterapia pozwala na redukcje masy guza i ułatwienie przeprowadzenia radykalnego zabiegu oraz zaoszczędzenie zwieraczy odbytu[460]. W kilku badaniach porównano skuteczność radioterapii w leczeniu raka odbytnicy przed operacją i po operacji[461][462]. Badanie CAO/ARO/AIO-94 wskazuje, że przedoperacyjna radioterapia wiązała się z niższym 10-letnim ryzykiem wznowy wynoszącym 7% w porównaniu do 10% dla leczenia pooperacyjnego, jednak nie stwierdzono różnicy w przeżyciu całkowitym[463][461]. Z kolei analiza danych SEER na chorych z guzem T3 N0 wskazuje, że radioterapia pooperacyjna jest związana z wydłużeniem przeżycia chorych, a radioterapia przedoperacyjna nie wpływa znacząco na przeżycie[464]. Napromieniowuje się lożę po guzie z 2–5 cm marginesem oraz okoliczne węzły chłonne, w tym przedkrzyżowe i biodrowe wewnętrzne. Zwykle łączna dawka wynosi 45–50 Gy w 25–28 dawkach frakcjonowanych. Po chemioradioterapii konieczna jest przerwa przed zabiegiem operacyjnym[465].
Chemioradioterapia jest stosowana w leczeniu neoadiuwantowym przed radykalną operacją w raku odbytnicy w zaawansowaniu T3–T4 N0 M0 lub T1–T4 N1–N2 M0 albo w leczeniu adiuwantowym w następstwie patomorfologicznej zmiany oceny stopnia zaawansowania na pT3 lub pN1–N2[368]. Korzyści wynikające z dodania chemioterapii do radioterapii wynikają z nasilenia toksyczności stosowanego promieniowania dla komórek nowotworowych i likwidacji mikroprzerzutów[460].
W badaniu klinicznym porównano skuteczność samodzielnej przedoperacyjnej (neoadiuwantowej) radioterapii z przedoperacyjną chemioradioterapią z użyciem 5-fluorouracylu z leukoworyną. Chemioradioterapia prowadziła do wyższego odsetka odpowiedzi na leczenie i niższego ryzyka wznowy, kosztem wyższej toksyczności. Nie zaobserwowano różnicy w przeżyciu całkowitym (OS) pomiędzy przedoperacyjną chemioradioterapią i samodzielną przedoperacyjną radioterapią[466]. W innym badaniu oceniono skuteczność chemioterapii przedoperacyjnej i pooperacyjnej do leczenia raka odbytnicy. Wykazano, że przedoperacyjna chemioterapia prowadzi do zmniejszenia masy guza i zmniejszenia zaawansowania zajęcia węzłów chłonnych[467]. Nie obserwowano różnicy w przeżyciu całkowitym leczonych przedoperacyjną i pooperacyjną chemioterapią[468]. Również w dwóch przeglądach systematycznych i jednaj metaanalizie stwierdzono, że dodanie chemioterapii do przedoperacyjnej radioterapii zmniejsza ryzyko nawrotu bez wpływu na przeżycie całkowite[469][470][471].
Wykazano, że kapecytabina jest równoważna 5-fluorouracylowi w leczeniu okołooperacyjnym raka odbytnicy. W badaniu NSABP R-04 porównującego leczenie okołooperacyjne za pomocą 5-fluorouracylu z lub bez oksaliplatyny lub kapecytabiny z lub bez oksaliplatyny nie stwierdzono różnicy w odsetku całkowitej odpowiedzi ocenianej histopatologicznie, odsetka zachowania zwieraczy odbytu i zmniejszenia zaawansowania[472]. W innym badaniu stwierdzono, że kapecytabina w porównaniu do 5-fluorouracylu wywołuje nie gorszy odsetek pięcioletnich przeżyć całkowitych i wykazuje pewną przewagę w wywoływaniu przeżycia wolnego od progresji choroby (DFS)[473]. W kilku badaniach wykazano, że dodanie oksaliplatyny do 5-fluorouracylu nie poprawia wyników leczenia[474][472]. W innym badaniu pomimo niższego odsetka występowania rezydualnej choroby podczas obserwacji nie zaobserwowano zwiększonego przeżycia wolnego od choroby i przeżycia całkowitego[475]. Podobnie dodanie oksaliplatyny do kapecytabiny w przedoperacyjnej chemioradioterapii nie prowadzi do zwiększenia przeżycia całkowitego[476].
Alternatywnym sposobem przedoperacyjnego leczenia raka odbytnicy jest krótkotrwałe napromienianie (ang. short-course preoperative radiotherapy), polegające na podawaniu 25 Gy w pięciu frakcjach po 5 Gy w ciągu kolejnych pięciu dni bez podawania chemioterapii[391][465]. Metoda jest często stosowana w Europie, w tym w Polsce, ze względu na większy komfort chorego i niższy koszt leczenia[391]. W randomizowanym polskim badaniu na 312 chorych porównano krótkotrwałą radioterapię z konwencjonalną chemioradioterapią. Zaobserwowano, że konwencjonalna chemioradioterapia nie zwiększa stopnia miejscowej kontroli choroby ani przeżycia całkowitego chorych. Chemioradioterapia wiązała się z większą toksycznością krótkoterminową[477]. Również w badaniu TROG nie stwierdzono częstości nawrotów oraz różnic w odsetku przeżyć całkowitych[478][479]. Inne badanie wskazuje, że choć długa chemioradioterapia w porównaniu do krótkotrwałej słabiej wpływa na zmniejszenie masy guza, to jednak nie ma różnicy w odsetku uzyskiwanych marginesów mikroskopowo ujemnych od nacieku nowotworowego[480].
Chemioterapia indukcyjna jest jedną z opcji leczenia okołooperacyjnego. Potencjalne korzyści w chemioterapii indukcyjnej obejmują likwidację mikroprzerzutów, wyższy odsetek odpowiedzi ocenianej histopatologicznie, ułatwienie operacji i poprawę tolerancji chemioterapii[481]. W kilku badaniach oceniono skuteczność chemioterapii indukcyjnej przeprowadzanej przed chemioradioterapią przedooperacyjną i operacją raka odbytnicy. W małym badaniu wykazano, że dodanie chemioterapii indukcyjnej za pomocą programu CapeOX składającego się z kapecytabiny i oksaliplatyny osiągnięto podobny odsetek odpowiedzi przy mniejszej toksyczności leczenia[482]. Inne badanie wykazało podobną skuteczność w leczeniu adiuwantowym 5-fluorouracylu i FOLFOX[483].
Adiuwantowa chemioterapia jest wskazana u wszystkich chorych w stadium II i III po neoadiuwantowej chemioradioterapii i radykalnym zabiegu chirurgicznym, jeśli nie otrzymali wcześniej chemioterapii neoadiuwantowej[484]. Niewiele badań oceniło skuteczność tego postępowania i jego rola nie jest w pełni zdefiniowana[484]. W wytycznych NCCN w leczeniu adiuwantowym w raku odbytnicy zaleca się stosowanie programu FOLFOX lub CapeOX jako preferowane metody leczenia. Stosowane mogą być również 5-fluorouracyl z leukoworyną, program FLOX lub kapecytabina[485]. W badaniu klinicznym wykazano, że adiuwantowa chemioterapia z użyciem 5-fluorouracylu po chemioterapii neoadiuwantowej nie redukowała ryzyka wznowy ani nie zwiększa odsetka przeżyć całkowitych (OS), jednak zwiększała odsetek przeżyć wolnych od choroby (DFS)[468]. W długoterminowej obserwacji w ramach tego badania również nie zaobserwowano wydłużenia przeżycia całkowitego, a wydłużenie przeżycia wolnego od choroby[486][484]. W metaanalizie na łącznie 9785 chorych z rakiem odbytnicy wykazano, że dodanie 5-fluorouracylu do leczenia pooperacyjnego wydłuża przeżycie całkowite (OS) i przeżycie wolne od choroby (DFS)[487]. W badaniu klinicznym ADORE porównano leczenie adiuwantowe za pomocą 5-fluorouracylu i programu FOLFOX. Wykazano, że program FOLFOX wywołuje wyższy odsetek przeżyć wolnych od choroby wynoszący 71,6% w porównaniu do leczonych 5-fluorouracylem, którzy osiągnęli 62,9% odsetek przeżyć wolnych od choroby[488].
Leczenie nawrotu choroby po operacji radykalnej onkologicznie
Nawrót miejscowy lub pojawienie się przerzutów odległych po radykalnej onkologicznie operacji dotyczy 15–40% chorych[489][490][491]. Nawrót częściej pojawia się w przypadku raka odbytnicy niż raka w okrężnicy, co jest związane z większą trudnością uzyskania odpowiedniego marginesu resekcji. Guzy odbytnicy wykazują tendencje do nawrotu miejscowego, a guzy okrężnicy do utworzenia przerzutów odległych[489][492]. Leczenie adiuwantowe zmniejsza ryzyko nawrotu o około 40%[491] i w ośmioletniej obserwacji 35% chorych doświadczyło nawrotu choroby nowotworowej[491]. Odsetek nawrotów po 1 roku od leczenia wynosił 12%, po dwóch latach od leczenia 14%, po trzech 8%, a po czterech i pięciu latach od leczenia odpowiednio 8% i 3%[493], 80% nawrotów pojawia się w ciągu pierwszych trzech lat od operacji[493]. Ryzyko nawrotu jest znacząco większe, gdy stwierdza się margines chirurgiczny z obecnym naciekiem nowotworowym (resekcje R1 i R2). W badaniu obserwacyjnym stwierdzono 40% ryzyko nawrotu choroby po resekcji z naciekiem nowotworowym marginesu resekcji, podczas gdy u chorych z ujemnym marginesem resekcji ryzyko nawrotu wynosiło 10%[490].
W przypadku miejscowego nawrotu w części przypadków możliwe jest wykonanie powtórnej operacji, co poprawia wyniki lokalnej kontroli choroby i może poprawiać przeżycie całkowite leczonych[494]. Celem reoperacji jest usunięcie nawrotowego guza wraz z marginesem zdrowych tkanek, a operacja powinna być połączona z leczeniem adiuwantowym. Zwykle uzyskanie pożądanych marginesów zdrowych tkanek jest bardzo trudne, co wymaga rozległego zakresu zabiegu. Decyzja o wykonaniu powtórnej operacji jest bardzo trudna, ponieważ może być oparta jedynie o pośrednie przesłanki wskazujące na nawrót obejmujące wzrost stężenia CEA, stwierdzenie zwiększonego wychwytu znacznika w PET czy niewielkie zmiany widoczne w tomografii komputerowej[489].
Leczenie nawrotu w postaci przerzutu odległego jest leczone na tych samych zasadach co choroba z przerzutami[495].
Leczenie choroby z przerzutami
Około 50–60% chorych z rakiem jelita grubego rozwija przerzuty odległe[450]. Podstawą leczenia choroby z przerzutami jest chemioterapia. U niewielkiej części chorych przerzuty mogą być usunięte operacyjnie, a mała grupa chorych z początkowo technicznie nieoperacyjnymi pojedynczymi przerzutami będzie mogła zostać zakwalifikowana do zabiegu po chemioterapii. Leczenie różni się w zależności od przyjętej optymalnej strategii leczenia choroby zaawansowanej klinicznie i chorzy w zależności od założonych celów leczenia mogą być podzieleni na cztery grupy kliniczne[496]:
- chorzy z pierwotnie operacyjnymi przerzutami do wątroby lub płuc bez przeciwwskazań do zabiegu – u chorych z pojedynczymi, korzystnie położonymi przerzutami wykonuje się ich resekcje z zachowaniem odpowiednio marginesu zdrowych tkanek. Zabieg jest uzupełniony adiuwantową chemioterapią lub poprzedzony neoadiuwantową chemioterapią[497].
- chorzy z potencjalnie operacyjnymi przerzutami – celem leczenia jest zmniejszenie wielkości przerzutów celem umożliwienia ewentualnej operacji i długotrwałego przeżycia chorych. W leczeniu wykorzystuje się najaktywniejsze intensywne programy chemioterapii z dwoma lub trzema lekami z lub bez użycia leków celowanych[372].
- chorzy z uogólnioną chorobą lub przerzutami ze zbyt małym prawdopodobieństwem wykonania udanej resekcji – celem leczenia jest maksymalne wydłużenie przeżycia chorych, w tym celu stosuje się intensywną chemioterapię za pomocą połączenia dwóch lub trzech leków z lub bez leków celowanych. U chorych odpowiadających na leczenie można powtórnie rozważyć kwalifikację do leczenia zabiegowego pojedynczych przerzutów[372].
- chorzy z chorobą uogólnioną leczeni paliatywnie – podstawowym celem leczenia jest złagodzenie objawów choroby, poprawa jakości życia i hamowanie progresji choroby, aby maksymalnie wydłużyć przeżycie leczonych. Stosuje się mniej toksyczne programy lecznicze za pomocą pojedynczych leków lub ich kombinacji[372].
Leczenie ogólnoustrojowe wydłuża przeżycie całkowite leczonych. Wybór leczenia zależy od stanu sprawności chorego, współistniejących chorób, występowania objawów choroby nowotworowej, szybkości progresji choroby i wcześniejszego leczenia[13]. U chorych w dobrym stanie sprawności stosuje się chemioterapię opartą o fluoropirymidynę (5-fluorouracyl, kapecytabina) w połączeniu z oksaliplatyną lub irynotekanem[15]. Wykorzystuje się programy wielolekowe: FOLFOX, CapeOX (XELOX), FOLFIRI i FOLFOXIRI. Chemioterapia może być kojarzona z inhibitorami EGFR (cetuksymab, panitumumab) lub inhibitorami angiogenezy (bewacyzumab)[373].
Przewaga wielolekowej chemioterapii w wydłużeniu przeżycia całkowitego jest mniej wyraźna i przeżycie zależy przede wszystkim od samego zastosowania aktywnych w chorobie leków, a w mniejszy sposób od ich kojarzenia w programy wielolekowe. Jednak programy wielolekowe w większym odsetku wywołują odpowiedź obiektywną i bardziej wydłużają przeżycie wolne od progresji choroby. U chorych starszych, ze współistniejącymi istotnymi chorobami, w gorszym stanie sprawności, z mniejszą dynamiką wzrostu choroby uzasadnione jest zastosowanie terapii sekwencyjnej z przynajmniej dwoma rzutami leczenia, którą charakteryzuje mniejsza toksyczność[13][15]. Optymalny czas trwania chemioterapii jest kwestią sporną. Typowo leczenie kontynuuje się do czasu wystąpienia progresji lub pojawienia się nieakceptowalnej toksyczności. Opcją jest także czasowe przerwanie intensywnego leczenia wielolekowego lub przejście na mniej intensywne leczenie podtrzymujące w momencie uzyskania kontroli i stabilizacji zmian nowotworowych, a następnie powtórne wprowadzenie leczenia skojarzonego w momencie progresji choroby[498]. Leczenie podtrzymujące po czasowym przerwaniu intensywnej chemioterapii wielolekowej jest złożone z 5-fluorouracylu z lub bez bewacyzumabu[372]. Ważna jest ciągła kontynuacja opieki nad chorymi celem zapewnienia i realizacji najlepszej strategii leczenia[499].
Leczenie potencjalnie operacyjnych przerzutów
- Metastazektomia
.jpg)
Metastazektomia jest to operacja wycięcia przerzutów. Starannie wybrani chorzy z rakiem jelita grubego z pojedynczymi, korzystnie zlokalizowanymi przerzutami do wątroby mogą być leczeni operacyjnie w celu poprawy rokowania. Chorzy z nieoperacyjnymi przerzutami lub nieresekcyjnym guzem pierwotnym nie kwalifikują się do metastazektomii. Na podobnych zasadach mogą być leczone przerzuty do płuc[500].
Operacja może być wykonana jednocześnie z wycięciem guza pierwotnego, a następnie uzupełniona chemioterapią adiuwantową lub jednoetapowa operacja jest poprzedzona chemioterapią neoadiuwantową lub wycięcie guza pierwotnego uzupełnia się leczeniem adiuwantowym, a następnie chorego poddaje się operacji usunięcia przerzutów[501]. Wykonuje się operacje nieanatomiczne (resekcje klinowe), hemihepatektomie i segmentektomie. W przypadku nacieku na istotne naczynie może być konieczna jego resekcja z odtworzeniem[502].
Warunkiem przeprowadzenia operacji jest możliwość resekcji przerzutów z uzyskaniem ujemnych mikroskopowo marginesów resekcji (resekcja R0) oraz utrzymanie koniecznych funkcji metabolicznych przez pozostały miąższ wątroby[503]. Konieczne jest zachowanie 1 cm marginesu resekcji[504]. Nie przeprowadza się metastazektomii w przypadku obecności nieresekowalnych przerzutów, ryzyka wystąpienia niewydolności wątroby po operacji, obecności niekontrolowanej choroby pozawątrobowej, szerokiego zajęcia węzłów chłonnych i obecności przerzutów do mózgu lub kości[504][505]. Paliatywna operacja wycięcia przerzutów jest rzadko wskazana i kluczowe w ich leczeniu jest leczenie ogólnoustrojowe[500].
Leczenie przerzutów do wątroby poprawia rokowanie. Mediana odsetka pięcioletniego przeżycia całkowitego leczonych resekcją przerzutów w różnych badaniach wynosi 20–40%[506][507][508][509]. Jednak chorzy z pojedynczym przerzutem do wątroby osiągają lepsze wyniki i badania wskazują na medianę pięcioletniego przeżycia całkowitego wynoszącą 70%[510][511][512][509].
Podobne zasady leczenia obowiązują w przypadku przerzutów do płuc. Warunkiem operacji jest założenie radykalnej resekcji mikroskopowo wolnej od nacieku nowotworu (resekcja R0), stan chorego pozwala na zabieg oraz nie występują przerzuty pozapłucne, ewentualnie mogą być obecne, ale muszą być kontrolowane za pomocą leczenia chirurgicznego lub innej formy terapii[513]. Nie ma randomizowanych badań oceniających skuteczność chirurgicznego leczenia przerzutów w płucach[514]. Niektóre badania sugerują, że korzyści przynosi jednoczesne chirurgiczne leczenie przerzutów w wątrobie i pozawątrobowych[515][516][517][518].
- Leczenie potencjalnie resekowalnych nieoperacyjnych przerzutów (konwersja do operacyjności)
Większość chorych z przerzutami nie kwalifikuje się do operacji. Część chorych z izolowanymi przerzutami do wątroby lub płuc nie kwalifikuje się do zabiegu z powodu nacieku ważnych struktur i może ona odnieść korzyści z chemioterapii, która może wywołać regresję przerzutów przywracając ich techniczną operacyjność. Chorzy z dużą liczbą przerzutów w wątrobie lub płucach nie kwalifikują się do operacji poprzedzonej chemioterapią, ponieważ szansa na uzyskanie radykalnej resekcji przerzutów jest niewielka, a sama chemioterapia rzadko pozwala na całkowite zniszczenie przerzutu. W próbie konwersji do zmian operacyjnych może być użyty każdy aktywny schemat stosowany w leczeniu choroby uogólnionej, ponieważ głównym celem nie jest likwidacja mikroprzerzutów, lecz uzyskanie regresji „widocznych” przerzutów[519].
W leczeniu stosuje się program FOLFIRI z lub bez bewacyzumabu, pogram FOLFOX z lub bez bewacyzumabu, program CapeOX z lub bez bewacyzumabu, program FOLFIRI z lub bez panitumumabu albo cetuksymabu, program FOLFOX z lub bez panitumumabu albo cetuksymabu, program FOLFOXIRI z lub bez bewacyzumabu[520].
W badaniu II fazy oceniono przydatność programu FOLFOX w leczeniu nieoperacyjnych przerzutów do wątroby. Wykazano, że u 60% leczonych objętość guza uległa zmniejszeniu i u 40% z wszystkich leczonych chemioterapia umożliwiła radykalny onkologicznie zabieg[521]. W badaniu na 1104 chorych z nieoperacyjnymi przerzutami do wątroby leczonych chemioterapią opartej głównie na oksaliplatynie leczenie u 12,5% chorych pozwoliło wykonać wtórną resekcję przerzutów[522].
W dwóch badaniach porównano skuteczność programów FOLFOX i FOLFOXIRI (5-fluorouracyl, leukoworyna, oksaliplatyna i irynotekan) w leczeniu chorych z nieoperacyjnymi przerzutami. W obu badaniach u chorych leczonych FOLFOXIRI zaobserwowano wzrost odsetka uzyskanych radykalnych resekcji w porównaniu z FOLFOX, w badaniu włoskim z 6% do 15%[523], a w badaniu greckim z 4% do 10%[524][525]. W badaniu greckim ponadto zaobserwowano wyższą medianę przeżycia całkowitego leczonych schematem FOLFOXIRI wynoszącą 24,4 miesiąca w porównaniu z medianą przeżycia leczonych programem FOLFOX wynoszącą 16,7 miesiąca[525].
W kilku badaniach oceniono skuteczność dodania inhibitora EGFR do schematu FOLFOXIRI lub FOLFOX w konwersji choroby nieoperacyjnej do operacyjnej. W badaniu CELIM porównano połączenie cetuksymabem z programem FOLFOXIRI lub FOLFOX. Zaobserwowano, że dodanie cetuksymabu wywołuje wzrost odsetka wykonanych resekcji z 32% do 60%[526]. Nie zaobserwowano różnicy przeżycia całkowitego pomiędzy grupami chorych leczonych cetuksymabem ze schematem FOLFOXIRI lub cetuksymabem z programem FOLFOX[527]. Również w innym badaniu porównującym połączenie cetuksymabu z programem FOLFOXIRI lub FOLFOX u chorych z nieoperacyjnymi przerzutami do wątroby dodanie cetuksymabu do chemioterapii prowadziło do większego odsetka uzyskanych resekcji, ponadto przeżycie chorych otrzymujących cetuksymab było wyższe niż chorych leczonych bez tego leku[528]. Metaanaliza czterech badań wykazała, że dodanie cetuksymabu lub panitumumabu do chemioterapii znacząco zwiększa przeżycie całkowite chorych i odsetek wykonanych resekcji[529].
Rola bewacyzumabu w konwersji nieoperacyjnych przerzutów do operacyjnych nie jest jasna. Badania sugerują, że dodanie bewacyzumabu do schematów opartych na irynotekanie zwiększa odsetek odpowiedzi na leczenie[530][531]. Z drugiej strony duże badanie wskazuje na brak korzyści dodania bewacyzumabu do chemioterapii[532].
- Chemioterapia neoadiuwantowa i adiuwantowa operacyjnych przerzutów
U wszystkich chorych z chorobą przerzutową (stadium IV) zaleca się stosowanie leczenia ogólnoustrojowego, które może zostać wdrożone przed operacją lub po zabiegu[533]. Programy lecznicze nie różnią się od stosowanych w leczeniu choroby zaawansowanej i przerzutowej. Sekwencja leczenia farmakologicznego i operacyjnego jest niejasna. Potencjalnie chemioterapia neoadiuwantowa pozwala na wcześniejsze leczenie mikroprzerzutów i określenie wrażliwości na leczenie. Wadą postępowania jest ryzyko progresji w trakcie leczenia uniemożliwiające leczenie operacyjne i, paradoksalnie, uzyskanie pełnej odpowiedzi utrudniającej ocenę ustalenia zakresu operacji[533].
Leczenie choroby z nieoperacyjnymi przerzutami

Leczenie nieoperacyjnej choroby z przerzutami jest oparte o chemioterapię. Chemioterapia na tym etapie zaawansowania choroby nie umożliwia wyleczenia, jednak poprzez kontrolę wzrostu guza i objawów choroby prowadzi do przedłużenia przeżycia chorych oraz przedłużenia czasu do progresji choroby. Wybór leczenia jest uwarunkowany założonymi celami leczenia, stanem sprawności chorego, obecnością współistniejących chorób i profilem toksyczności terapii[534]. Programy leczenia mogą być złożone z jednego leku lub kombinacji kilku leków[535]. W leczeniu pierwszego rzutu u chorych kwalifikujących się do intensywnego leczenia stosuje się jeden z pięciu schematów wielolekowych: program FOLFOX z lub bez bewacyzumabu albo cetuksymabu/panitumumabu[b], program CapeOX, program FOLFIRI z lub bez bewacyzumabu albo cetuksymabu/panitumumabu[b], program FOLFOXIRI z lub bez bewacyzumabu i 5-fluorouracyl z leukoworyną albo kapecytabinę[536][535]. Badania porównujące skuteczność leczenia pierwszej linii w intensywnej chemioterapii nie wykazały znaczącej przewagi żadnego z tych pięciu programów leczenia i żaden z nich nie jest preferowany wobec pozostałych programów pierwszej linii[537].
- FOLFOX
Program FOLFOX jest połączeniem 5-fluorouracylu, leukoworyny i oksaliplatyny[538]. Schemat FOLFOX może być stosowany samodzielnie lub w połączeniu z bewacyzumabem lub w połączeniu z inhibitorami EGFR (cetyksymab, panitumumab) w leczeniu pierwszego rzutu u chorych z zaawansowaną chorobą w ramach intensywnej chemioterapii[535][539].
W badaniu III fazy na około 200 chorych porównano leczenie za pomocą 5-fluorouracylu z leukoworyną z lub bez dodania oksaliplatyny. Zaobserwowano, że w grupie chorych otrzymujących chemioterapię z dodaniem oksaliplatyny wywołuje ona wyższy odsetek odpowiedzi obiektywnych wynoszący 53% w porównaniu do 16% oraz wyższą medianę przeżycia wolnego od progresji wynoszącą w grupie leczonych 5-fluorouracylem z oksaliplatyną 8,7 miesiąca i 7,4 miesiąca w grupie bez oksaliplatyny[540]. Połączenia 5-fluorouracylu, leukoworyny i oksaliplatyny w schemat FOLFOX1 i 2 nigdy nie oceniono w badaniach z randomizacją[541]. Programy FOLFOX3 i FOLFOX4 w badaniu klinicznym wykazały skuteczność u chorych z progresją po leczeniu 5-fluorouracylem. Program FOLFOX4 wywołał nieco wyższy odsetek odpowiedzi i niższy stopień toksyczności niż FOLFOX3[542][541]. W innym badaniu porównano skuteczność schematu LV5FU2 (5-fluorouracyl, leukoworyna) z programem FOLFOX4 w leczeniu wcześniej nie leczonych chorych z przerzutowym rakiem jelita grubego. Wykazano, że chorzy leczeni schematem FOLFOX4 wykazywali wyższy odsetek odpowiedzi obiektywnych niż leczeni LV5FU2, który wynosił odpowiednio 51% i 22%, a także wyższą medianę czasu wolnego od progresji (PFS) wynoszącą 9 miesięcy dla chorych leczonych FOLFOX4 w porównaniu do 6,2 miesiąca u leczonych LV5FU2. Przewaga przeżycia całkowitego u leczonych FOLFOX4 nie osiągnęła istotności statystycznej[543].
W badaniu EORTC 40983 oceniono skuteczność chemioterapii FOLFOX zastosowanej okołooperacyjnie u chorych zakwalifikowanych do resekcji przerzutów do wątroby. Wykazano wzrost odsetka przeżyć wolnych od choroby (DFS). Nie zaobserwowano różnicy w przeżyciu całkowitym (OS) pomiędzy leczonych wyłącznie chirurgicznie i leczonych chirurgicznie z chemioterapią w programie FOLFOX, choć mogło być to spowodowane przez częstsze stosowanie chemioterapii II-rzutu w grupie leczonych wyłącznie chirurgicznie[544][545][546]
Schemat FOLFOX stosowany bez połączenia z lekami celowanymi nie ma przewagi nad programem CapeOX (XELOX)[547] i oba programy mogą być stosowane zamiennie[539].
- CapeOX (XELOX)
Schemat CapeOX (XELOX) jest połączeniem kapecytabiny i oksaliplatyny[538]. Może być stosowany w leczeniu pierwszego rzutu u chorych z chorobą zaawansowaną w ramach intensywnej chemioterapii[548]. Do programu CapeOX może zostać dodany bewacyzumab[539].
W dużym badaniu III fazy porównano skuteczność leczenia za pomocą programu FOLFOX oraz CapeOX u chorych z przerzutowym rakiem jelita grubego i zaobserwowano, że leczenie programem CapeOX i FOLFOX wywołuje podobne mediany przeżycia wolnego od progresji (PFS) oraz podobne mediany przeżycia całkowitego (OS)[549][550][551]. W metaanalizie 7 badań na łącznie 3603 chorych z przerzutowym rakiem jelita grubego wykazano, że u leczonych programami FOLFOX i CapeOX mediana czasu przeżycia wolnego od progresji (PFS) i przeżycia całkowitego (OS) nie różnią się statystycznie[552].
- FOLFIRI
Schemat FOLFIRI jest połączeniem 5-fluorouracylu, leukoworyny i irynotekanu[553]. Schemat FOLFIRI może być stosowany samodzielnie lub w połączeniu z bewacyzumabem lub w połączeniu z inhibitorami EGFR (cetyksymab, panitumumab) w leczeniu pierwszego rzutu u chorych z chorobą zaawansowaną w ramach intensywnej chemioterapii[554]. Program FOLFIRI może być łączony z bewacyzumabem lub inhibitorami EGFR[555].
W badaniu klinicznym porównano skuteczność programu FOLFIRI i FOLFOX6, chorych losowo przydzielano do leczenia programem FOLFOX lub FOLFIRI, a po wystąpieniu progresji podawano drugi badany schemat jako leczenie drugiej linii. Oba programy wykazały dość podobne wyniki. Program FOLFOX wywołał odpowiedź u 54% leczonych, mediana przeżycia wolnego od progresji (PFS) wynosiła 8,0 miesięcy, z kolei schemat FOLFIRI wywoływał odpowiedź obiektywną u 56% leczonych, a mediana przeżycia wolnego od progresji wynosiła 8,5 miesiąca[556]. Podobnie w kolejnym badaniu porównującym skuteczność programów FOLFIRI i FOLFOX nie zaobserwowano różnicy pomiędzy odsetkami odpowiedzi obiektywnych, czasem wolnym od progresji choroby (DFS) i medianami przeżycia całkowitego (OS)[557].
- FOLFOXIRI
Schemat FOLFOXIRI jest połączeniem 5-fluorouracylu, leukoworyny, oksaliplatyny i irynotekanu[558]. Schemat FOLFOXIRI może być stosowany samodzielnie lub w połączeniu z bewacyzumabem w leczeniu pierwszego rzutu u chorych z chorobą zaawansowaną w ramach intensywnej chemioterapii[559].
W badaniu GONO u 244 chorych z zaawansowanym rakiem jelita grubego porównano leczenie schematem FOLFOXIRI i FOLFOX. Zaobserwowano, że leczeni programem FOLFOXIRI osiągnęli wyższą medianę przeżycia wolnego od progresji choroby wynoszącą 9,8 miesiąca w porównaniu do 6,9 miesiąca u leczonych programem FOLFOX oraz wyższą medianę przeżycia całkowitego wynoszącą 22,6 miesiąca w porównaniu do 16,7 miesiąca dla leczonych FOLFOX[523][525]. Z kolei w badaniu HORG nie stwierdzono istotnej statystycznie różnicy przeżycia całkowitego pomiędzy leczonymi programem FOLFOXIRI i FOLFOX[524]. Program FOLFOXIRI wykazuje większą toksyczność niż FOLFOX[555].
- 5-fluorouracyl z leukoworyną lub kapecytabina
Połączenie 5-fluorouracylu z leukoworyną, monoterapia kapecytabiną, połączenie 5-fluorouracylu z leukoworyną i bewacyzumabem i połączenie kapecytabiny z bewacyzumabem mogą być stosowane w leczeniu pierwszego rzutu u chorych ze słabszą tolerancją bardziej agresywnego i toksycznego leczenia[559][555].
W analizie dwóch badań porównujących samodzielne leczenie chirurgiczne przerzutów z zabiegiem uzupełnionym chemioterapią za pomocą 5-fluorouracylu z leukoworyną wykazano, że adiuwantowa chemioterapia po wycięciu przerzutów wydłuża przeżycie wolne od progresji choroby (PFS), którego mediana u chorych leczonych adiuwantową chemioterapią wynosiła 27,9 miesiąca, a wyłącznie metastazektomią 18,8 miesiąca[560]. Wykazano, że dodanie bewacyzumabu do kapecytabiny wydłuża przeżycie wolne od progresji choroby (PFS) w porównaniu do monoterapii kapecytabiną[561].
- Bewacyzumab
Bewacyzumab jest przeciwciałem monoklonalnym wiążącym się z czynnikiem wzrostu śródbłonka naczyniowego (VEGF), co hamuje tworzenie naczyń stymulowane przez rozrastający się nowotwór i uzależniony od angiogenezy wzrost guza i przerzutów[562]. Bewacyzumab dodany do chemioterapii pierwszej linii oferuje umiarkowane korzyści kliniczne[563]. Może być dodany do programu FOLFOX, CapeOX (XELOX), FOLFIRI, FOLFOXIRI, 5-fluorouracylu z leukoworyną lub kapecytabiny[536]. Nie zaleca się stosowania bewacyzumabu w leczeniu uzupełniającym po resekcji przerzutów[563].
Kilka badań wskazuje, że dodanie bewacyzumabu do 5-fluorouracylu i leukoworyny wydłuża przeżycie całkowite (OS) leczonych[531][564][565]. W analizie kilku badań wykazano, że dodanie bewacyzumabu do 5-fluorouracylu i leukoworyny wydłuża medianę przeżycia całkowitego. Chorzy otrzymujący leczenie z bewacyzumabem osiągnęli medianę przeżycia całkowitego wynoszącą 17,9 miesiąca, a chorzy leczeni bez bewacyzumabu 14,6 miesiąca[566]. Z kolei w badaniu na 1400 chorych z przerzutowym rakiem jelita grubego oceniającym skuteczność dodania bewacyzumabu do programów leczniczych opartych na oksaliplatynie (FOLFOX4 i CapeOX) zaobserwowano wydłużenie przeżycia wolnego od progresji choroby (PFS) o 1,4 miesiąca, ale nie stwierdzono istotnego statystycznie wydłużenia przeżycia całkowitego (OS)[532]. Nie ma randomizowanych badań porównujących skuteczność programu FOLFIRI z lub bez bewacyzumabu[567][568]. Kilka metaanaliz wskazuje, że dodanie bewacyzumabu do chemioterapii powoduje wydłużenie mediany przeżycia wolnego od progresji choroby (PFS) i niewielkie wydłużenie mediany przeżycia całkowitego[569][570][563][571][572][573].
Dodanie bewacyzumabu do chemioterapii wiąże się z wyższą śmiertelnością związaną z leczeniem[574]. Początkowe badania przedkliniczne sugerowały, że przerwanie leczenia bewacyzumabem może spowodować przyspieszenie nawrotu połączonego z jego większą agresywnością. Późniejsze badania kliniczne nie potwierdziły istnienia efektu z odbicia[575][576].
- Inhibitory EGFR
Cetuksymab i panitumumab są przeciwciałami monoklonalnymi skierowanymi przeciw receptorowi naskórkowego czynnika wzrostu (EGF), który aktywuje szlaki MAPK/ERK i PI3K/AKT/mTOR[229][243], które z kolei zwiększają zdolność do proliferacji, chronią komórki przed apoptozą, ułatwiają migrację komórek i promują angiogenezę[229][212]. Inhibitory EGFR mogą być stosowane w leczeniu skojarzonym z programami FOLFOX i FOLFIRI pierwszego rzutu u chorych z chorobą zaawansowaną w ramach intensywnej chemioterapii[577]. Inhibitory EGFR lub bewacyzumabu dodane do programów chemioterapii pierwszej linii są równoważnymi opcjami terapeutycznymi[578].
Cetuksymab i panitumumab wykazują skuteczność tylko u 10–20% chorych[579][580][581], mimo że nadmierną ekspresję receptora EGF stwierdza się w 25–80% przypadków raka jelita grubego[244][243][582][583]. Jednocześnie ocena nadekspresji EGFR nie pozwala przewidzieć skuteczności leczenia[584][579][585][580][586], dlatego nie zaleca się jej rutynowego oznaczania, a ujemny wynik badania nie dyskwalifikuje chorego z leczenia tymi lekami[581]. Zaleca się oznaczenie mutacji KRAS i NRAS, ponieważ chorzy z rozpoznaną mutacją KRAS lub NRAS nie odnoszą korzyści z leczenia inhibitorami EGFR[581]. Również niektóre mutacje BRAF powodują oporność na leczenie inhibitorami EGFR i zaleca się oznaczenie mutacji BRAF, choć wartość predykcyjna oznaczenia jest niepewna[587].
Skuteczność cetuksymabu dodanego do programu FOLFOX oceniono w kilku badaniach klinicznych. W badaniu OPUS wykazano, że dodanie cetuksymabu do programu FOLFOX zwiększa odsetek odpowiedzi obiektywnych oraz wydłuża czas przeżycia wolnego od progresji (PFS). Nie zaobserwowano wydłużenia mediany przeżycia całkowitego[588]. Z kolei w badaniu MRC COIN nie potwierdzono korzyści w wydłużeniu czasu przeżycia wolnego od progresji (PFS) po dodaniu cetuksymabu do schematu FOLFOX lub CapeOX[589]. Jednak analiza zbiorcza wyników tego badania i badania OPUS wskazuje na korzyści w odsetku odpowiedzi obiektywnych i czasu wolnego od progresji choroby (PFS)[590]. W dużym badaniu porównano leczenie schematem FOLFOX z cetyksymabem z leczeniem programem FOLFOX z bewacyzumabem i wykazano, że przeżycie całkowite jest zbliżone w obu metodach leczenia[591].
W badaniu CRYSTAL zbadano skuteczność połączenia cetuksymabu z programem FOLFIRI w porównaniu do schematu FOLFIRI stosowanego bez kojarzenia z inhibitorami EGFR. U chorych leczonych cetuksymabem z FOLFIRI stwierdzono wydłużenie przeżycia wolnego od progresji choroby oraz przeżycia całkowitego w stosunku do FOLFIRI stosowanego samodzielnie[592][249]. Jednak połączenie nie poprawia jakości życia leczonych[593]. W innym badaniu porównano skuteczność leczenia za pomocą panitumumabu i programu FOLFIRI z leczeniem za pomocą FOLFOX z bewacyzumabem. Zaobserwowano, że panitumumab z FOLFIRI wywołuje lepszą poprawę przeżycia całkowitego niż bewacyzumab z FOLFIRI[594].
Skuteczność panitumumabu w połączeniu z programem FOLFOX oceniono w dużym randomizowanym badaniu. Zaobserwowano, że dodanie panitumumabu do schematu FOLFOX wywołuje wydłużenie przeżycia wolnego od progresji (PFS) oraz wydłużenie przeżycia całkowitego (OS)[595]. W małym badaniu oceniającym skuteczność leczenia za pomocą panitumumabu z FOLFOX i bewacyzumabu z FOLFOX stwierdzono poprawę przeżycia całkowitego w grupie leczonych za pomocą panitumumabu z FOLFOX[596].
Skuteczność panitumumabu w połączeniu z programem FOLFIRI oceniono jedynie jako leczenie drugiej linii. W kilku badaniach stwierdzono wydłużenie czasu wolnego od progresji[248][597][598], a w jednym badaniu zaobserwowano wydłużenie przeżycia wolnego od progresji choroby[599].
- Leczenie II linii
Leczenie po progresji choroby zależy od zastosowanego leczenia pierwszej linii[578]:
- chorzy, którzy w pierwszej linii leczenia byli leczeni programem FOLFOX lub CapeOX (XELOX) w drugiej linii terapii mogą być leczeni za pomocą programu FOLFIRI lub irynotekanu stosowanej samodzielnie lub w połączeniach z lekami celowanymi: bewacyzumabem, cetuksymabem[b], panitumumabem[b], ramucirumabem lub afliberceptem. Ze względu na koszty i profil toksyczności preferowanym lekiem anty-VEGF jest bewacyzumab[600],
- chorzy, którzy w pierwszej linii leczenia byli leczeni programem FOLFIRI w drugiej linii terapii mogą być leczeni za pomocą programu FOLFOX lub CapeOX (XELOX) podawanych samodzielnie lub w połączeniu z bewacyzumabem, irynotekanem w połączeniu z cetuksymabem lub panitumumabem[b], ewentualnie cetuksymabem lub panitumumabem w monoterapii[b],
- chorzy, którzy w pierwszej linii leczenia byli leczeni za pomocą 5-fluorouracylu lub kapecytabiny bez połączenia z oksaliplatyną lub irynotekanem w drugiej linii terapii mogą być leczeni za pomocą programu FOLFOX, CapeOX, FOLFIRI, irynotekanu w monoterapii lub irynotekanu z oksaliplatyną (IRINOX),
- chorzy, którzy w pierwszej linii leczenia byli leczeni programem FOLFOXIRI w drugiej linii terapii mogą być leczeni za pomocą połączenia irynotekanu z cetuksymabem lub panitumumabem[b], monoterapii cetuksymabem lub panitumumabem[b].
Irynotekan w porównaniu do 5-fluorouracylu bardziej wydłuża przeżycie całkowite (OS) i przeżycie wolne od progresji choroby (PFS)[601]. Nie stwierdzono różnicy przeżycia całkowitego pomiędzy chorymi leczonymi irynotekanem w monoterapii lub programem FOLFOX[602]. Metaanaliza wykazała, że dodanie leku celowanego do klasycznej chemioterapii poprawia wyniki leczenia kosztem wyższej toksyczności[603].
Panitumumab w leczeniu drugiej linii w monoterapii poprawia przeżycie wolne od progresji choroby w porównaniu do najlepszej terapii wspomagającej[247][580]. Panitumumab dodany do programu FOLFIRI wydłuża przeżycie wolne od progresji choroby w stosunku do samodzielnego leczenia za pomocą FOLFIRI[248][604]. Cetuksymab w leczeniu drugiej linii w monoterapii[605][246][579] lub w połączeniu z irynotekanem wydłuża przeżycie wolne od progresji choroby[579][606].
W badaniu ML18147 bewacyzumab dodany do chemioterapii II linii przedłużał przeżycie wolne od progresji choroby i nieznacznie wpływał na wydłużenie przeżycia całkowitego[607][608], podobnie w badaniu BEBYP bewacyzumab wydłużał przeżycie wolne od progresji choroby[609]. Kontynuacja leczenia bewacyzumabem mimo progresji po leczeniu połączeniem chemioterapii I linii i bewacyzumabu może poprawiać przeżycie całkowite[610].
Aflibercept jest przeciwciałem wiążącym się z czynnikiem wzrostu śródbłonka naczyniowego (VEGF)[611]. W badaniu VELOUR aflibercept dodany do programu FOLFIRI wydłużał przeżycie wolne od progresji oraz przeżycie całkowite[612]. Lek wykazuje aktywność tylko w połączeniu z programem FOLFIRI u chorych nie leczonych wcześniej za pomocą tego schematu[600].
Ramucirumab jest przeciwciałem skierowanym przeciw receptorowi VEGFR2[613]. W badaniu RAISE wykazano, że dodanie ramucirumabu do schematu FOLFIRI powoduje wydłużenie przeżycia całkowitego w stosunku do leczenia za pomocą wyłącznie programu FOLFIRI[614].
Leczenie paliatywne i wspomagające
Leczenie paliatywne jest to terapia ukierunkowana na poprawę jakości życia poprzez walkę z uciążliwymi objawami choroby nowotworowej.
- Niedrożność jelita grubego

.jpg)
Niedrożność jelita pojawia się w 10–25% przypadków raka jelita grubego[615][616][617][618]. Niedrożność częściej pojawia się w guzach esicy i w okolicy zgięcia śledzionowego, co jest następstwem ich fizjologicznie węższego światła[616][619]. Szacuje się, że niedrożność jest spowodowana guzem zlokalizowanym w 11% przypadków w kątnicy, wstępnicy w 5% przypadków, zgięcia wątrobowego w 3% przypadków, poprzecznicy w 11% przypadków, zgięcia śledzionowego w 12% przypadków, zstępnicy w 10% przypadków, esicy w 35% przypadków i odbytnicy w 13% przypadków[620]. Niedrożność w raku jelita grubego zwykle jest efektem stopniowego zarastania światła narządu[616]. Perforacja z powodu narastającego obrzęku i martwicy ściany jelita towarzyszy 3–8% przypadków niedrożności[620]. Niedrożność jelita grubego pogarsza rokowanie[621].
Typ i nasilenie objawów zależą od progresji od niedrożności przepuszczającej do pełnej niedrożności. Typowo objawy mają charakter powolnie narastający, a dolegliwości mają trudny do określenia początek[622]. Niepełna niedrożność objawia się bólem brzucha, wzdęciami, zaparciami ze zmniejszeniem kalibru stolca, możliwe są naprzemienne biegunki i zaparcia[17][623]. W późniejszym etapie pojawia się pełna niedrożność, w której występuje kolkowy, a później również tępy ból brzucha, nudności i wymioty, wzdęcia, zatrzymanie stolca i gazów. W badaniu fizykalnym bywa możliwe palpacyjne stwierdzenie obecności guza[617][622]. W związku z rozwijającymi się zaburzeniami wodno-elektrolitowymi, gospodarki kwasowo-zasadowej oraz narastającej toksemii pojawiają się objawy ogólnoustrojowe, które nieleczone ostatecznie prowadzą do wstrząsu i zgonu[285].
Niedrożność jelita grubego z powodu nowotworu jest rozpoznawana na podstawie objawów klinicznych, badania fizykalnego oraz badań obrazowych[617][285].
Leczenie niedrożności z powodu nowotworu złośliwego jest bardzo trudne, szczególnie gdy jest ona pierwszym objawem nierozpoznanej wcześniej choroby nowotworowej. Na wybór najlepszego postępowania wpływa dotychczasowy przebieg choroby, uzyskana odpowiedź na leczenie i stan ogólny chorego[617][622]. W pierwszym etapie leczenia wyrównuje się zaburzenia wodno-elektrolitowe i kwasowo-zasadowe poprzez odpowiednio intensywne dożylne leczenie płynami. Konieczne jest założenie sondy nosowo-żołądkowej i odsysanie treści jelitowej. Zmniejsza to rozdęcie jelit, nasilenie wymiotów, ilość bakterii i ich toksyn oraz zapobiega zachłystowemu zapaleniu płuc. Rozplem bakterii i rozwijającą się sepsę kontroluje się za pomocą antybiotyków o szerokim spektrum działania obejmującym również bakterie beztlenowe[285]. Sprzyja to ustąpieniu subniedrożności, co daje czas na wdrożenie diagnostyki, co może ułatwić skuteczniejsze leczenie[617].
U chorych w dobrym stanie ogólnym i jednym poziomem niedrożności może być wykonana operacja przywracająca drożność jelita[617]. Przeciwwskazaniem do leczenia operacyjnego jest niedrożność wielopoziomowa oraz niedrożność spowodowana masywnym wysiewem do otrzewnej[622]. U chorych w złym stanie ogólnym, w zaawansowanej chorobie czy starszym wieku wykonanie zabiegu może być niemożliwe, mimo technicznej możliwości jego wykonania[617].
Leczenie może być operacyjne, endoskopowe z założeniem stentu lub zachowawcze[617]. Leczenie operacyjne polega na jednoetapowej resekcji guza z pierwotnym odtworzeniem ciągłości przewodu pokarmowego lub operacji z wyłonieniem stomii, która w kolejnych etapach leczenia może być zamknięta po odtworzeniu ciągłości przewodu pokarmowego[624][615][625]. Operacja Hartmanna polega na usunięciu guza z wyłonieniem kolostomii w pierwszym etapie, a następnie odtworzeniu ciągłości przewodu pokarmowego w drugim etapie leczenia. Operacja polegająca na wyłonieniu stomii dwulufowej z usunięciem guza w drugim etapie leczenia i ewentualnym odtworzeniem ciągłości przewodu pokarmowego w trzecim etapie nie wykazuje przewagi nad operacją Hartmanna, która jest preferowana w sytuacjach klinicznych wymagających leczenia dwuetapowego[624]. Operacja Hartmanna nie wykazuje przewagi nad odcinkową resekcją jelita z jednoczasowym zespoleniem. Operacja Hartmanna prawdopodobnie nie oferuje korzyści w przeżyciu całkowitym, ale może być ona lepsza dla chorych z dużym ryzykiem operacyjnym[624].
Niedrożność może być leczona za pomocą mało inwazyjnej procedury endoskopowego założenia samorozprężalnego stentu. Stenty powodują poszerzenie światła jelita poprzez ściśnięcie guza. Metalowe elementy wywierają ucisk na guz powodując jego miejscową martwicę, co ułatwia jego zakotwiczenie. Na zakotwiczenie wpływa klepsydrowaty kształt stentu, co również utrudnia jego niepożądaną migrację i nawrót niedrożności[616][615]. Założenie stentu pozwala uniknąć operacji u nieuleczalnie chorych lub odroczyć ją u chorych wymagających pilnego zabiegu[616]. Endoskopowe założenie stentu w porównaniu do zabiegu chirurgicznego wykazuje mniejszą śmiertelność okołooperacyjną i mniejszą chorobowość okołooperacyjną[626] kosztem wyższego odsetka późnych powikłań[627]. Stentowanie w porównaniu do zabiegu chirurgicznego wykazuje mniejszą skuteczność w paliatywnym łagodzeniu objawów niedrożności[627], jednak zabieg operacyjny nie daje znaczącej przewagi w przeżyciu całkowitym[628][627][615]. Około 20% chorych wymaga powtórnego zabiegu z powodu zamknięcia światła przez nowotwór lub migracji stentu[629][616]. Stenty samorozprężalne nie są wskazane w przypadku guzów zlokalizowanych w kanale odbytu, ponieważ mogą wywołać uczucie bólu, parcia na stolec lub nietrzymanie moczu[615][616]. W guzach odbytnicy bywa stosowany laser Nd:YAG[616].
W przypadku nieoperacyjnej niedrożności jelita grubego z powodu nowotworu leczenie skupia się na łagodzeniu objawów choroby i zapewnieniu możliwie najlepszej jakości życia. Konieczne jest opanowanie bólu, nudności i wymiotów oraz ograniczenie ilości przyjmowanego pokarmu. Ból jest opanowywany za pomocą leków przeciwbólowych podawanych według drabiny analgetycznej. Ze względu na nudności i wymioty unika się doustnego podawania leków i preferuje się drogę dożylną i przezskórną. Preferowanym opioidem jest fentanyl. W leczeniu nudności i wymiotów stosuje się leki antycholinergiczne (hioscyna, skopolamina), metoklopramid, niektóre neuroleptyki (haloperydol, chloropromazyna), antagonisty 5-HT3 (ondansetron, granisetron i inne) oraz glikokortykosteroidy. Leki przeciwsekrecyjne zmniejszają wydzielanie z przewodu pokarmowego ograniczając nudności, wymioty i ból. Największe kliniczne znaczenie wykazuje hioscyna, skopolamina i oktreotyd (analog somatostatyny). W pełnej niedrożności w leczeniu paliatywnym lekiem przeciwwymiotnym z wyboru jest haloperydol[617].
U chorych, u których możliwe jest wykonanie zabiegu przywracającego drożność jelita podaje się całkowite żywienie pozajelitowe. Długoterminowe żywienie pozajelitowe chorych z guzem nieoperacyjnym jest kontrowersyjne ze względów etycznych i raczej nie jest rutynowo stosowane u tych chorych[630][631][617].
- Perforacja jelita
Perforacja jelita grubego dotyczy około 2,5–10% przypadków raka jelita grubego[281][282][283]. Może być ona efektem martwicy guza, martwicy wzdętego jelita, zwykle kątnicy, lub powikłaniem leczenia zabiegowego[632][616]. Perforacja pojawia się w zaawansowanym stadium choroby, w 53% przypadków występuje w IV stopniu zaawansowania klinicznego, w 37% przypadków w stadium IIIb i 12% w IIIa[281][633]. Stanowi stan zagrożenia życia z powodu rozwoju rozlanego zapalenia otrzewnej lub powstania ropnia wewnątrzotrzewnowego[616]. Powikłanie jest związane z wysoką śmiertelnością krótkoterminową osiągającą 20–40%[634] i ze złym rokowaniem długoterminowym[616][632].
Głównym objawem perforacji jelita jest bardzo silnym ból brzucha oraz objawy rozwijającego się zapalenia otrzewnej[616]. Leczenie jest operacyjne i jego podstawowym celem jest opanowanie rozlanego zapalenia otrzewnej i rozwijającej się sepsy. Zwykle wykonuje się operację Hartmanna z wycięciem fragmentu jelita obejmującego perforację z wyłonieniem tymczasowej kolostomii, konieczne jest płukanie jamy brzusznej[635][616].
- Krwawienie z guza

Krwawienie w raku jelita grubego najczęściej występuje pod postacią przewlekłą prowadzącą do niedokrwistości. Ostre i zagrażające życiu krwotoki z guza są raczej rzadkie, jednak wymagają pilnej operacji[634]. Krwawienie z guza może stanowić poważny problem w zaawansowanej chorobie nowotworowej, szczególnie u chorych ze wskazaniami do leczenia przeciwzakrzepowego[616]. Podstawą rozpoznania jest kolonoskopia, która umożliwia lokalizację miejsca krwawienia. Należy wykluczyć krwawienie z górnego odcinka przewodu pokarmowego. Pomocna może być selektywna angiografia lub scyntygrafia za pomocą znakowanych 99Tc krwinkami czerwonymi[636][285].
Celem leczenia jest usunięcie źródła krwawienia. Przed zabiegiem zaburzenia wodno-elektrolitowe są wyrównywane za pomocą leczenia płynami, a w niektórych sytuacjach może być konieczna transfuzja koncentratu krwinek czerwonych[285]. Jeśli jest to możliwe przeprowadza się radykalny onkologicznie zabieg. W przypadku choroby nieoperacyjnej wykonywana jest koagulacja plazmowa za pomocą beamera argonowego lub ablacja przy użyciu lasera Nd:YAG. Powodują one martwicę skrzepową lub waporyzację guza, ale zwykle konieczne jest kilkakrotne powtarzanie zabiegu. Koagulacja za pomocą argonu ze względu na niewielką penetrację odznacza się mniejszym ryzykiem perforacji jelita[615].
- Ból nowotworowy

Przewlekłych dolegliwości bólowych doświadcza około 30% chorych z rakiem jelita grubego[637]. Leczenie bólu nowotworowego jest oparte o leki przeciwbólowe. Leki o coraz wyższej sile działania są podawane w określonej kolejności ułożonej według drabiny analgetycznej. Początkowo w pierwszym stopniu leczenia podawane są niesteroidowe leki przeciwzapalne. Gdy okazują się one nieskuteczne mimo zwiększenia ich dawki, to leczenie bólu jest intensyfikowane i w drugim stopniu leczenia znajdują zastosowanie słabe opioidy takie jak kodeina, dihydrokodeina oraz tramadol. W przypadku ich nieskuteczności mimo zwiększenia dawki lub wystąpienia objawów niepożądanych podawane są silne opioidy obejmujące morfinę, oksykodon, fentanyl i buprenorfina. Preferuje się podawanie doustne lub przezskórne (w tym drugim przypadku za pomocą specjalnych plastrów). Leki doustne są podawane jako preparaty o zmodyfikowanym uwalnianiu służące do długotrwałej kontroli dolegliwości oraz jako preparaty o natychmiastowym uwalnianiu mające za zadanie doraźną kontrolę bólu. Silny ból może wymagać dożylnego podania leku. Ustalanie podstawowej dawki silnego opioidu rozpoczyna się od stosowania morfiny o natychmiastowym uwalnianiu, po czym przechodzi się na doustne opioidy o zmodyfikowanym uwalnianiu lub na plastry o działaniu przezskórnym. Kluczowe dla skuteczności leczenia przeciwbólowego jest stosowanie leków w regularnych odstępach czasu[638]. Pomocniczo wykorzystywane są koanalgetyki, które same nie są lekami przeciwbólowymi, ale wspierają działanie leków przeciwbólowych[639].
W uporczywych dolegliwościach bólowych korzysta się z chirurgicznych blokad splotów i nerwów. Neuroliza splotu podbrzusznego górnego jest wykonywana w bólu w środkowej części brzucha i odbytnicy[640][641][642][643]. Bywa wykonywana neuroliza splotu trzewnego[644][645][640]. W leczeniu bólu odbytnicy wykonuje się neurolizę splotu podbrzusznego górnego[640], zwoju nieparzystego[646][647][648] lub pośrodkową mielotomię[640].
- Zespół kacheksja-anoreksja
Zespół kacheksja-anoreksja jest rozpoznawany u 54% chorych na raka jelita grubego[649][650]. Jest to złożony zespół zaburzeń prowadzących do spadku masy ciała w połączeniu z utratą apetytu, osłabieniem, niedokrwistością i zaburzeniami odpowiedzi immunologicznej w następstwie zaburzeń metabolicznych spowodowanych chorobą nowotworową[651][652]. W patogenezie zespołu największą role odgrywają cytokiny prozapalne i neuroprzekaźniki[653]. Zespół kacheksja-anoreksja znacznie pogarsza jakość życia i prowadzi do wysokiej śmiertelności[654].
Nie ma ogólnie przyjętego sposobu leczenia kacheksji. W leczeniu stosuje się progestageny (octan megestrolu, medroksyprogesteron) oraz kortykosteroidy. Istotna jest odpowiednia dieta i ćwiczenia fizyczne dostosowane do stanu chorego[655]. Niesteroidowe leki przeciwzapalne mogą powodować przyrost masy ciała, poprawiać jakość życia i stan sprawności[655][656][654]. W badaniach klinicznych wykazano skuteczność ibuprofenu[657], indometacyny[658], celekoksybu[659] i rofekoksybu[660]. Jednak nie ma wystarczających dowodów na ich rutynowe stosowanie w leczeniu zespołu kacheksja-anoreksja[661]. Niektóre badania sugerują, że w leczeniu zespołu kacheksja-anoreksja mogą być skuteczne grelina[662], antagonisty melanokortyny[663][664], formoterol[665] i talidomid[666].
Rokowanie
Przeżycie chorych na raka jelita grubego silnie zależy od stopnia zaawansowania klinicznego. Odsetek przeżyć pięcioletnich w raku jelita grubego wynosi około 65–70%[667][668][669]. Pięć lat przeżywa 90% chorych z rakiem w zaawansowaniu miejscowym, 70% chorych w zaawansowaniu lokalnym i 10% chorych z chorobą uogólnioną[28][668].
| Stadium kliniczne | Przeżycie całkowite |
| I | 93% |
| IIa | 85% |
| IIb | 72% |
| IIIa | 83% |
| IIIb | 64% |
| IIIc | 44% |
| IV | 8% |
| Stadium kliniczne | Przeżycie całkowite w raku okrężnicy | Przeżycie całkowite w raku odbytnicy |
| I | 92% | 87% |
| IIa | 87% | 80%[c] |
| IIb | 63%[d] | 49%[c] |
| IIIa | 89%[d] | 84%[c] |
| IIIb | 69%[d] | 71% |
| IIIc | 53% | 58% |
| IV | 11% | 12% |
Historia

Początkowo leczenie raka jelita grubego było oparte wyłącznie na leczeniu chirurgicznym. Operacje raka jelita grubego były przez długi czas uważane za bardzo ryzykowne, ze względu na powikłania infekcyjne[671]. Dopiero wprowadzenie pierwszych sulfonamidów w latach 30. XX wieku pozwoliło znacznie obniżyć śmiertelność związaną z zabiegiem[672]. Jean-François Reybard w 1823 roku przeprowadził resekcje guza esicy z wykonaniem zespolenia[671]. W 1883 roku Karel Maydl wykonał pierwszą prawostronną kolektomię[671][673]. Po raz pierwszy pełna kolektomia została przeprowadzona przez René Leriche w 1914 roku[671].
W drugiej połowie XIX wieku guzy odbytnicy wycinano wyłącznie od strony krocza. Pierwszą operację usunięcia odbytnicy z dostępu kroczowego wykonał w 1826 roku Jacques Lisfranc[673][674]. Mimo wprowadzanych kolejnych modyfikacji operacji wprowadzanych przez Theodora Billrotha, Aristide Verneuil, Emila Kochera, Ludwika Rydygiera i Paula Kraske polegających na usuwaniu kości guzicznej, kości krzyżowej lub innych struktur operacje charakteryzowała wysoka śmiertelność z powodu powikłań septycznych oraz wysoki odsetek wczesnych nawrotów[675][676]. Ernest Miles w 1908 wykonał i opisał operację brzuszno-kroczową raka odbytnicy, jednak zabieg przeprowadził wcześniej w 1879 roku czeski chirurg Vincenz Czerny[671][675][673]. Technikę operacji brzuszno-kroczowej systematycznie poprawiano, co umożliwiło jej wykonywanie w jednym etapie[674]. W 1921 roku Henri Albert Hartmann do leczenia guzów w esicy i górnej części odbytnicy wprowadził dwuetapową operację z dostępu brzusznego. W 1939 C. F. Dixon przeprowadził operacje jednoetapową z jednoczesnym zespoleniem. Wówczas stosowano szeroki 5 cm dolny margines resekcji, co ograniczało możliwości zastosowania operacji zachowującej zwieracze w leczeniu raków położonych w odbytnicy[677]. Jednak to operacja brzuszno-kroczowa w pierwszej połowie XX wieku była złotym standardem dla leczenia guzów położonych w odbytnicy[674]. Dopiero badania wykazujące znacznie mniejszy konieczny margines resekcji umożliwiły operacje z zachowaniem zwieraczy również w guzach położonych w górnej i środkowej części odbytnicy[677]. Na przełomie lat 40. i 50. wykazano przewagę mniej okaleczającej przedniej resekcji[674]. W latach 70. XX wieku do chirurgii wprowadzono szew mechaniczny, co ułatwiło wykonywanie niskich przednich resekcji odbytnicy i pozwoliło na dalsze ograniczenie wskazań do operacji brzuszno-kroczowej[678]. W 1982 roku R.J. Heald wprowadził całkowite wycięcie mezorektum, co znacząco poprawiło wyniki operacji raka odbytnicy[679]. Na początku lat 90. do chirurgii raka jelita grubego wprowadzono operacje laparoskopowe[680].
Początki chemioterapii sięgają końca lat 50. W 1958 roku do leczenia zaawansowanego raka jelita grubego wprowadzono 5-fluorouracyl[681]. W 1975 wykazano, że 5-fluorouracyl połączony z radioterapią (chemioradioterapia) znacząco podwyższa odsetek odpowiedzi całkowitych[682][683]. Pierwsze badania nad leczeniem okołooperacyjnym raka jelita grubego prowadzono w latach 70. W połowie lat 80. standardem leczenia uzupełniającego stał się 5-fluorouracyl[684][685]. W przełomie lat 80. i 90. do 5-fluorouracylu w celu zmniejszania toksyczności zaczęto dołączać leukoworynę[686][685]. Na początku lat dwutysięcznych wprowadzono irynotekan[687] i oksaliplatynę, które stały się podstawą współczesnych programów wielolekowych[688].
Choroba u zwierząt
Złośliwe nabłonkowe nowotwory jelita nie są częste wśród zwierząt, szczególnie w porównaniu do raka jelita grubego u ludzi, u których jest bardzo częstym nowotworem. U zwierząt rak znacznie częściej pojawia się w obrębie jelita cienkiego niż grubego[689]. Rak jelita grubego został stwierdzony u różnych grup taksonomicznych zwierząt, w tym u niektórych gadów[690][691] i ssaków, w tym gryzoni[692][693], bydła[689], koni[689][694], psów[695][689], kotów[692][696] i niektórych naczelnych (tamaryna białoczuba)[692]. U psów częściej rak jest zlokalizowany w odbytnicy niż okrężnicy[695].
Rak jelita grubego u zwierząt objawia się obecnością świeżej krwi w kale, bolesnym parciem i uporczywymi próbami defekacji. Mogą występować wymioty, utrata apetytu i spadek masy ciała. W badaniu fizykalnym bywa stwierdzana obecność guza w jamie brzusznej, bolesność podczas palpacji, wodobrzusze i obecność guza w badaniu przez odbyt[696][694].
W rozpoznaniu choroby wykorzystuje się kontrastowe badanie radiologiczne, endoskopię i ultrasonografię. W badaniach laboratoryjnych stwierdza się niedokrwistość. Pomocna w rozpoznaniu bywa biopsja cienkoigłowa podejrzanej zmiany. Ostateczne rozpoznanie jest stawiane na podstawie badania histopatologicznego[696][695].
Leczenie jest oparte o chirurgiczną resekcję zmiany[696][695]. Sporadycznie bywa stosowana radioterapia[696][697]. Chemioterapii nie stosuje się rutynowo[697].
Uwagi
Przypisy
- ↑ Krzakowski i in. 2015 ↓, s. 611.
- ↑ Krzakowski i in. 2015 ↓, s. 635.
- ↑ a b World Cancer Research Fund International: Worldwide data. [dostęp 2015-10-26]. [zarchiwizowane z tego adresu (2015-10-26)].
- ↑ a b c Labianca i in. 2013 ↓, s. 1.
- ↑ a b J. Ferlay, I. Soerjomataram, R. Dikshit, S. Eser i inni. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. „Int J Cancer”. 136 (5), s. E359-86, marzec 2015. DOI: 10.1002/ijc.29210. PMID: 25220842.
- ↑ a b c d e f g h i Krzakowski i in. 2015 ↓, s. 614.
- ↑ Krzakowski i in. 2015 ↓, s. 612.
- ↑ a b Krzakowski i in. 2015 ↓, s. 614–615.
- ↑ a b c Krzakowski i in. 2015 ↓, s. 616.
- ↑ a b c d Krzakowski i in. 2015 ↓, s. 617.
- ↑ Krzakowski i in. 2015 ↓, s. 630–631.
- ↑ a b Krzakowski i in. 2015 ↓, s. 631–634.
- ↑ a b c d Krzakowski i in. 2015 ↓, s. 620.
- ↑ Krzakowski i in. 2015 ↓, s. 618–619.
- ↑ a b c d e Roman Herman, Jarosław Reguła, Jakub Pałucki, Wojciech Polkowski, Piotr Potemski: Rak okrężnicy. 2014.
- ↑ a b c d Szczeklik i Gajewski 2014 ↓, s. 974.
- ↑ a b c d e f Podolsky i in. 2015 ↓, s. 1568.
- ↑ a b c d Bailey i in. 2012 ↓, s. 234.
- ↑ a b Kordek 2007 ↓, s. 181.
- ↑ H.S. Saidi, D. Karuri, E.O. Nyaim. Correlation of clinical data, anatomical site and disease stage in colorectal cancer. „East Afr Med J”. 85 (6), s. 259–262, czerwiec 2008. PMID: 18817021.
- ↑ a b S.R. Majumdar, R.H. Fletcher, A.T. Evans. How does colorectal cancer present? Symptoms, duration, and clues to location. „Am J Gastroenterol”. 94 (10), s. 3039–3045, październik 1999. DOI: 10.1111/j.1572-0241.1999.01454.x. PMID: 10520866.
- ↑ Kordek 2007 ↓, s. 180.
- ↑ Kordek 2007 ↓, s. 180–181.
- ↑ a b Swan 2006 ↓, s. 67.
- ↑ a b c Podolsky i in. 2015 ↓, s. 1569.
- ↑ Ho 2012 ↓, s. 76.
- ↑ a b C.M. Johnson, C. Wei, J.E. Ensor, D.J. Smolenski i inni. Meta-analyses of colorectal cancer risk factors. „Cancer Causes Control”. 24 (6), s. 1207–1222, czerwiec 2013. DOI: 10.1007/s10552-013-0201-5. PMID: 23563998.
- ↑ a b c d e f g h i j k F.A. Haggar, R.P. Boushey. Colorectal cancer epidemiology: incidence, mortality, survival, and risk factors. „Clin Colon Rectal Surg”. 22 (4), s. 191–197, listopad 2009. DOI: 10.1055/s-0029-1242458. PMID: 21037809.
- ↑ P. Boyle, J.S. Langman. ABC of colorectal cancer: Epidemiology. „BMJ”. 321 (7264), s. 805–808, wrzesień 2000. PMID: 11009523.
- ↑ a b c d e f g h i j k l m n M. Pericleous, D. Mandair, M.E. Caplin. Diet and supplements and their impact on colorectal cancer. „J Gastrointest Oncol”. 4 (4), s. 409–423, grudzień 2013. DOI: 10.3978/j.issn.2078-6891.2013.003. PMID: 24294513.
- ↑ a b c d e f g h i Lin 2009 ↓, s. 361–372.
- ↑ T. Norat, A. Lukanova, P. Ferrari, E. Riboli. Meat consumption and colorectal cancer risk: dose-response meta-analysis of epidemiological studies. „Int J Cancer”. 98 (2), s. 241–256, marzec 2002. PMID: 11857415.
- ↑ M.S. Sandhu, I.R. White, K. McPherson. Systematic review of the prospective cohort studies on meat consumption and colorectal cancer risk: a meta-analytical approach. „Cancer Epidemiol Biomarkers Prev”. 10 (5), s. 439–446, maj 2001. PMID: 11352852.
- ↑ R. Sinha, W.H. Chow, M. Kulldorff, J. Denobile i inni. Well-done, grilled red meat increases the risk of colorectal adenomas. „Cancer Res”. 59 (17), s. 4320–4324, wrzesień 1999. PMID: 10485479.
- ↑ N.M. Probst-Hensch, R. Sinha, M.P. Longnecker, J.S. Witte i inni. Meat preparation and colorectal adenomas in a large sigmoidoscopy-based case-control study in California (United States). „Cancer Causes Control”. 8 (2), s. 175–183, marzec 1997. PMID: 9134241.
- ↑ D.D. Alexander, A.J. Miller, C.A. Cushing, K.A. Lowe. Processed meat and colorectal cancer: a quantitative review of prospective epidemiologic studies. „Eur J Cancer Prev”. 19 (5), s. 328–341, wrzesień 2010. DOI: 10.1097/CEJ.0b013e32833b48fa. PMID: 20495462.
- ↑ D.D. Alexander, C.A. Cushing. Red meat and colorectal cancer: a critical summary of prospective epidemiologic studies. „Obes Rev”. 12 (5), s. e472–e493, maj 2011. DOI: 10.1111/j.1467-789X.2010.00785.x. PMID: 20663065.
- ↑ S. Franceschi, C. La Vecchia, A. Russo, A. Favero i inni. Macronutrient intake and risk of colorectal cancer in Italy. „Int J Cancer”. 76 (3), s. 321–324, maj 1998. PMID: 9579566.
- ↑ a b W.C. Willett, M.J. Stampfer, G.A. Colditz, B.A. Rosner i inni. Relation of meat, fat, and fiber intake to the risk of colon cancer in a prospective study among women. „N Engl J Med”. 323 (24), s. 1664–1672, grudzień 1990. DOI: 10.1056/NEJM199012133232404. PMID: 2172820.
- ↑ T.T. Fung, F.B. Hu, K. Wu, S.E. Chiuve i inni. The Mediterranean and Dietary Approaches to Stop Hypertension (DASH) diets and colorectal cancer. „Am J Clin Nutr”. 92 (6), s. 1429–1435, grudzień 2010. DOI: 10.3945/ajcn.2010.29242. PMID: 21097651.
- ↑ D.D. Alexander, C.A. Cushing, K.A. Lowe, B. Sceurman i inni. Meta-analysis of animal fat or animal protein intake and colorectal cancer. „Am J Clin Nutr”. 89 (5), s. 1402–1409, maj 2009. DOI: 10.3945/ajcn.2008.26838. PMID: 19261724.
- ↑ S. Sasazuki, M. Inoue, M. Iwasaki, N. Sawada i inni. Intake of n-3 and n-6 polyunsaturated fatty acids and development of colorectal cancer by subsite: Japan Public Health Center-based prospective study. „Int J Cancer”. 129 (7), s. 1718–1729, październik 2011. DOI: 10.1002/ijc.25802. PMID: 21120874.
- ↑ Y. Kimura, S. Kono, K. Toyomura, J. Nagano i inni. Meat, fish and fat intake in relation to subsite-specific risk of colorectal cancer: The Fukuoka Colorectal Cancer Study. „Cancer Sci”. 98 (4), s. 590–597, kwiecień 2007. DOI: 10.1111/j.1349-7006.2007.00425.x. PMID: 17425596.
- ↑ E. Theodoratou, G. McNeill, R. Cetnarskyj, S.M. Farrington i inni. Dietary fatty acids and colorectal cancer: a case-control study. „Am J Epidemiol”. 166 (2), s. 181–195, lipiec 2007. DOI: 10.1093/aje/kwm063. PMID: 17493949.
- ↑ S. Kim, D.P. Sandler, J. Galanko, C. Martin i inni. Intake of polyunsaturated fatty acids and distal large bowel cancer risk in whites and African Americans. „Am J Epidemiol”. 171 (9), s. 969–979, maj 2010. DOI: 10.1093/aje/kwq032. PMID: 20392864.
- ↑ S. Wu, B. Feng, K. Li, X. Zhu i inni. Fish consumption and colorectal cancer risk in humans: a systematic review and meta-analysis. „Am J Med”. 125 (6), s. 551–559.e5, czerwiec 2012. DOI: 10.1016/j.amjmed.2012.01.022. PMID: 22513196.
- ↑ C.R. Daniel, M.L. McCullough, R.C. Patel, E.J. Jacobs i inni. Dietary intake of omega-6 and omega-3 fatty acids and risk of colorectal cancer in a prospective cohort of U.S. men and women. „Cancer Epidemiol Biomarkers Prev”. 18 (2), s. 516–525, luty 2009. DOI: 10.1158/1055-9965.EPI-08-0750. PMID: 19190143.
- ↑ X.J. Shen, J.D. Zhou, J.Y. Dong, W.Q. Ding i inni. Dietary intake of n-3 fatty acids and colorectal cancer risk: a meta-analysis of data from 489 000 individuals. „Br J Nutr”. 108 (9), s. 1550–1556, listopad 2012. DOI: 10.1017/S0007114512003546. PMID: 22906228.
- ↑ C.H. MacLean, S.J. Newberry, W.A. Mojica, P. Khanna i inni. Effects of omega-3 fatty acids on cancer risk: a systematic review. „JAMA”. 295 (4), s. 403–415, styczeń 2006. DOI: 10.1001/jama.295.4.403. PMID: 16434631.
- ↑ F.J. van Duijnhoven, H.B. Bueno-De-Mesquita, P. Ferrari, M. Jenab i inni. Fruit, vegetables, and colorectal cancer risk: the European Prospective Investigation into Cancer and Nutrition. „Am J Clin Nutr”. 89 (5), s. 1441–1452, maj 2009. DOI: 10.3945/ajcn.2008.27120. PMID: 19339391.
- ↑ A.E. Millen, A.F. Subar, B.I. Graubard, U. Peters i inni. Fruit and vegetable intake and prevalence of colorectal adenoma in a cancer screening trial. „Am J Clin Nutr”. 86 (6), s. 1754–1764, grudzień 2007. PMID: 18065596.
- ↑ K.B. Michels, E. Giovannucci, A.T. Chan, R. Singhania i inni. Fruit and vegetable consumption and colorectal adenomas in the Nurses’ Health Study. „Cancer Res”. 66 (7), s. 3942–3953, kwiecień 2006. DOI: 10.1158/0008-5472.CAN-05-3637. PMID: 16585224.
- ↑ J.S. Witte, M.P. Longnecker, C.L. Bird, E.R. Lee i inni. Relation of vegetable, fruit, and grain consumption to colorectal adenomatous polyps. „Am J Epidemiol”. 144 (11), s. 1015–1025, grudzień 1996. PMID: 8942431.
- ↑ D.S. Alberts, M.E. Martínez, D.J. Roe, J.M. Guillén-Rodríguez i inni. Lack of effect of a high-fiber cereal supplement on the recurrence of colorectal adenomas. Phoenix Colon Cancer Prevention Physicians’ Network. „N Engl J Med”. 342 (16), s. 1156–1162, kwiecień 2000. DOI: 10.1056/NEJM200004203421602. PMID: 10770980.
- ↑ E. Bidoli, S. Franceschi, R. Talamini, S. Barra i inni. Food consumption and cancer of the colon and rectum in north-eastern Italy. „Int J Cancer”. 50 (2), s. 223–229, styczeń 1992. PMID: 1730516.
- ↑ G.R. Howe, E. Benito, R. Castelleto, J. Cornée i inni. Dietary intake of fiber and decreased risk of cancers of the colon and rectum: evidence from the combined analysis of 13 case-control studies. „J Natl Cancer Inst”. 84 (24), s. 1887–1896, grudzień 1992. PMID: 1334153.
- ↑ B. Trock, E. Lanza, P. Greenwald. Dietary fiber, vegetables, and colon cancer: critical review and meta-analyses of the epidemiologic evidence. „J Natl Cancer Inst”. 82 (8), s. 650–661, kwiecień 1990. PMID: 2157027.
- ↑ Y. Park, D.J. Hunter, D. Spiegelman, L. Bergkvist i inni. Dietary fiber intake and risk of colorectal cancer: a pooled analysis of prospective cohort studies. „JAMA”. 294 (22), s. 2849–2857, grudzień 2005. DOI: 10.1001/jama.294.22.2849. PMID: 16352792.
- ↑ D. Aune, D.S. Chan, R. Lau, R. Vieira i inni. Dietary fibre, whole grains, and risk of colorectal cancer: systematic review and dose-response meta-analysis of prospective studies. „BMJ”. 343, s. d6617, 2011. PMID: 22074852.
- ↑ C.F. Garland, F.C. Garland. Do sunlight and vitamin D reduce the likelihood of colon cancer?. „Int J Epidemiol”. 9 (3), s. 227–231, wrzesień 1980. PMID: 7440046.
- ↑ I. Kato, A. Akhmedkhanov, K. Koenig, P.G. Toniolo i inni. Prospective study of diet and female colorectal cancer: the New York University Women’s Health Study. „Nutr Cancer”. 28 (3), s. 276–281, 1997. DOI: 10.1080/01635589709514588. PMID: 9343837.
- ↑ E.D. Gorham, C.F. Garland, F.C. Garland, W.B. Grant i inni. Vitamin D and prevention of colorectal cancer. „J Steroid Biochem Mol Biol”. 97 (1–2), s. 179–194, październik 2005. DOI: 10.1016/j.jsbmb.2005.06.018. PMID: 16236494.
- ↑ F. Levi, C. Pasche, F. Lucchini, C. La Vecchia. Selected micronutrients and colorectal cancer. a case-control study from the canton of Vaud, Switzerland. „Eur J Cancer”. 36 (16), s. 2115–2119, październik 2000. PMID: 11044650.
- ↑ J. Wactawski-Wende, J.M. Kotchen, G.L. Anderson, A.R. Assaf i inni. Calcium plus vitamin D supplementation and the risk of colorectal cancer. „N Engl J Med”. 354 (7), s. 684–696, luty 2006. DOI: 10.1056/NEJMoa055222. PMID: 16481636.
- ↑ M.L. McCullough, A.S. Robertson, C. Rodriguez, E.J. Jacobs i inni. Calcium, vitamin D, dairy products, and risk of colorectal cancer in the Cancer Prevention Study II Nutrition Cohort (United States). „Cancer Causes Control”. 14 (1), s. 1–12, luty 2003. PMID: 12708719.
- ↑ P. Pietinen, N. Malila, M. Virtanen, T.J. Hartman i inni. Diet and risk of colorectal cancer in a cohort of Finnish men. „Cancer Causes Control”. 10 (5), s. 387–396, październik 1999. PMID: 10530608.
- ↑ M.E. Martínez, E.L. Giovannucci, G.A. Colditz, M.J. Stampfer i inni. Calcium, vitamin D, and the occurrence of colorectal cancer among women. „J Natl Cancer Inst”. 88 (19), s. 1375–1382, październik 1996. PMID: 8827015.
- ↑ E. Kampman, R.A. Goldbohm, P.A. van den Brandt, P. van 't Veer. Fermented dairy products, calcium, and colorectal cancer in The Netherlands Cohort Study. „Cancer Res”. 54 (12), s. 3186–3190, czerwiec 1994. PMID: 8205538.
- ↑ E. Cho, S.A. Smith-Warner, D. Spiegelman, W.L. Beeson i inni. Dairy foods, calcium, and colorectal cancer: a pooled analysis of 10 cohort studies. „J Natl Cancer Inst”. 96 (13), s. 1015–1022, lipiec 2004. PMID: 15240785.
- ↑ A.A. Moghaddam, M. Woodward, R. Huxley. Obesity and risk of colorectal cancer: a meta-analysis of 31 studies with 70,000 events. „Cancer Epidemiol Biomarkers Prev”. 16 (12), s. 2533–2547, grudzień 2007. DOI: 10.1158/1055-9965.EPI-07-0708. PMID: 18086756.
- ↑ L.C. Thygesen, M. Grønbaek, C. Johansen, C.S. Fuchs i inni. Prospective weight change and colon cancer risk in male US health professionals. „Int J Cancer”. 123 (5), s. 1160–1165, wrzesień 2008. DOI: 10.1002/ijc.23612. PMID: 18546286.
- ↑ S.C. Larsson, A. Wolk. Obesity and colon and rectal cancer risk: a meta-analysis of prospective studies. „Am J Clin Nutr”. 86 (3), s. 556–565, wrzesień 2007. PMID: 17823417.
- ↑ N. Khan, F. Afaq, H. Mukhtar. Lifestyle as risk factor for cancer: Evidence from human studies. „Cancer Lett”. 293 (2), s. 133–143, lipiec 2010. DOI: 10.1016/j.canlet.2009.12.013. PMID: 20080335.
- ↑ Y. Kim, Y. Kim, S. Lee. An association between colonic adenoma and abdominal obesity: a cross-sectional study. „BMC Gastroenterol”. 9, s. 4, 2009. DOI: 10.1186/1471-230X-9-4. PMID: 19144203.
- ↑ S.H. Davoodi, T. Malek-Shahabi, A. Malekshahi-Moghadam, R. Shahbazi i inni. Obesity as an important risk factor for certain types of cancer. „Iran J Cancer Prev”. 6 (4), s. 186–194, 2013. PMID: 25250133.
- ↑ S.C. Larsson, N. Orsini, A. Wolk. Diabetes mellitus and risk of colorectal cancer: a meta-analysis. „J Natl Cancer Inst”. 97 (22), s. 1679–1687, listopad 2005. DOI: 10.1093/jnci/dji375. PMID: 16288121.
- ↑ L. Deng, Z. Gui, L. Zhao, J. Wang i inni. Diabetes mellitus and the incidence of colorectal cancer: an updated systematic review and meta-analysis. „Dig Dis Sci”. 57 (6), s. 1576–1585, czerwiec 2012. DOI: 10.1007/s10620-012-2055-1. PMID: 22350783.
- ↑ H. Yuhara, C. Steinmaus, S.E. Cohen, D.A. Corley i inni. Is diabetes mellitus an independent risk factor for colon cancer and rectal cancer?. „Am J Gastroenterol”. 106 (11), s. 1911–1921; quiz 1922, listopad 2011. DOI: 10.1038/ajg.2011.301. PMID: 21912438.
- ↑ a b S. Jarvandi, N.O. Davidson, M. Schootman. Increased risk of colorectal cancer in type 2 diabetes is independent of diet quality. „PLoS One”. 8 (9), s. e74616, 2013. DOI: 10.1371/journal.pone.0074616. PMID: 24069323.
- ↑ Y.X. Yang, S. Hennessy, J.D. Lewis. Type 2 diabetes mellitus and the risk of colorectal cancer. „Clin Gastroenterol Hepatol”. 3 (6), s. 587–594, czerwiec 2005. PMID: 15952101.
- ↑ J.M. Berster, B. Göke. Type 2 diabetes mellitus as risk factor for colorectal cancer. „Arch Physiol Biochem”. 114 (1), s. 84–98, luty 2008. DOI: 10.1080/13813450802008455. PMID: 18465362.
- ↑ K.T. Mills, C.F. Bellows, A.E. Hoffman, T.N. Kelly i inni. Diabetes mellitus and colorectal cancer prognosis: a meta-analysis. „Dis Colon Rectum”. 56 (11), s. 1304–1319, listopad 2013. DOI: 10.1097/DCR.0b013e3182a479f9. PMID: 24105007.
- ↑ Y. Mao, S. Pan, S.W. Wen, K.C. Johnson. Physical inactivity, energy intake, obesity and the risk of rectal cancer in Canada. „Int J Cancer”. 105 (6), s. 831–837, lipiec 2003. DOI: 10.1002/ijc.11159. PMID: 12767070.
- ↑ G.A. Colditz, C.C. Cannuscio, A.L. Frazier. Physical activity and reduced risk of colon cancer: implications for prevention. „Cancer Causes Control”. 8 (4), s. 649–667, lipiec 1997. PMID: 9242482.
- ↑ a b T.I. Nilsen, L.J. Vatten. Prospective study of colorectal cancer risk and physical activity, diabetes, blood glucose and BMI: exploring the hyperinsulinaemia hypothesis. „Br J Cancer”. 84 (3), s. 417–422, luty 2001. DOI: 10.1054/bjoc.2000.1582. PMID: 11161410.
- ↑ K.J. Lee, M. Inoue, T. Otani, M. Iwasaki i inni. Physical activity and risk of colorectal cancer in Japanese men and women: the Japan Public Health Center-based prospective study. „Cancer Causes Control”. 18 (2), s. 199–209, marzec 2007. DOI: 10.1007/s10552-006-0098-3. PMID: 17206529.
- ↑ A.L. Zisman, A. Nickolov, R.E. Brand, A. Gorchow i inni. Associations between the age at diagnosis and location of colorectal cancer and the use of alcohol and tobacco: implications for screening. „Arch Intern Med”. 166 (6), s. 629–634, marzec 2006. DOI: 10.1001/archinte.166.6.629. PMID: 16567601.
- ↑ E. Botteri, S. Iodice, V. Bagnardi, S. Raimondi i inni. Smoking and colorectal cancer: a meta-analysis. „JAMA”. 300 (23), s. 2765–2778, grudzień 2008. DOI: 10.1001/jama.2008.839. PMID: 19088354.
- ↑ a b P.S. Liang, T.Y. Chen, E. Giovannucci. Cigarette smoking and colorectal cancer incidence and mortality: systematic review and meta-analysis. „Int J Cancer”. 124 (10), s. 2406–2415, maj 2009. DOI: 10.1002/ijc.24191. PMID: 19142968.
- ↑ M. Lüchtenborg, K.K. White, L. Wilkens, L.N. Kolonel i inni. Smoking and colorectal cancer: different effects by type of cigarettes?. „Cancer Epidemiol Biomarkers Prev”. 16 (7), s. 1341–1347, lipiec 2007. DOI: 10.1158/1055-9965.EPI-06-0519. PMID: 17626999.
- ↑ E. Botteri, S. Iodice, S. Raimondi, P. Maisonneuve i inni. Cigarette smoking and adenomatous polyps: a meta-analysis. „Gastroenterology”. 134 (2), s. 388–395, luty 2008. DOI: 10.1053/j.gastro.2007.11.007. PMID: 18242207.
- ↑ a b L.M. Hannan, E.J. Jacobs, M.J. Thun. The association between cigarette smoking and risk of colorectal cancer in a large prospective cohort from the United States. „Cancer Epidemiol Biomarkers Prev”. 18 (12), s. 3362–3367, grudzień 2009. DOI: 10.1158/1055-9965.EPI-09-0661. PMID: 19959683.
- ↑ a b c d e f E.R. Kim, D.K. Chang. Colorectal cancer in inflammatory bowel disease: the risk, pathogenesis, prevention and diagnosis. „World J Gastroenterol”. 20 (29), s. 9872–9881, sierpień 2014. DOI: 10.3748/wjg.v20.i29.9872. PMID: 25110418.
- ↑ a b J.A. Eaden, K.R. Abrams, J.F. Mayberry. The risk of colorectal cancer in ulcerative colitis: a meta-analysis. „Gut”. 48 (4), s. 526–535, kwiecień 2001. PMID: 11247898.
- ↑ a b L. Lakatos, G. Mester, Z. Erdelyi, G. David i inni. Risk factors for ulcerative colitis-associated colorectal cancer in a Hungarian cohort of patients with ulcerative colitis: results of a population-based study. „Inflamm Bowel Dis”. 12 (3), s. 205–211, marzec 2006. DOI: 10.1097/01.MIB.0000217770.21261.ce. PMID: 16534422.
- ↑ C. Canavan, K.R. Abrams, J. Mayberry. Meta-analysis: colorectal and small bowel cancer risk in patients with Crohn’s disease. „Aliment Pharmacol Ther”. 23 (8), s. 1097–1104, kwiecień 2006. DOI: 10.1111/j.1365-2036.2006.02854.x. PMID: 16611269.
- ↑ A. Ekbom, C. Helmick, M. Zack, H.O. Adami. Ulcerative colitis and colorectal cancer. A population-based study. „N Engl J Med”. 323 (18), s. 1228–1233, listopad 1990. DOI: 10.1056/NEJM199011013231802. PMID: 2215606.
- ↑ J. Potack, S.H. Itzkowitz. Colorectal cancer in inflammatory bowel disease. „Gut Liver”. 2 (2), s. 61–73, wrzesień 2008. DOI: 10.5009/gnl.2008.2.2.61. PMID: 20485613.
- ↑ R.M. Soetikno, O.S. Lin, P.A. Heidenreich, H.S. Young i inni. Increased risk of colorectal neoplasia in patients with primary sclerosing cholangitis and ulcerative colitis: a meta-analysis. „Gastrointest Endosc”. 56 (1), s. 48–54, lipiec 2002. PMID: 12085034.
- ↑ K.W. Nuako, D.A. Ahlquist, D.W. Mahoney, D.J. Schaid i inni. Familial predisposition for colorectal cancer in chronic ulcerative colitis: a case-control study. „Gastroenterology”. 115 (5), s. 1079–1083, listopad 1998. PMID: 9797361.
- ↑ J. Askling, P.W. Dickman, P. Karlén, O. Broström i inni. Family history as a risk factor for colorectal cancer in inflammatory bowel disease. „Gastroenterology”. 120 (6), s. 1356–1362, maj 2001. PMID: 11313305.
- ↑ P. Ferrari, M. Jenab, T. Norat, A. Moskal i inni. Lifetime and baseline alcohol intake and risk of colon and rectal cancers in the European prospective investigation into cancer and nutrition (EPIC). „Int J Cancer”. 121 (9), s. 2065–2072, listopad 2007. DOI: 10.1002/ijc.22966. PMID: 17640039.
- ↑ T. Mizoue, M. Inoue, K. Wakai, C. Nagata i inni. Alcohol drinking and colorectal cancer in Japanese: a pooled analysis of results from five cohort studies. „Am J Epidemiol”. 167 (12), s. 1397–1406, czerwiec 2008. DOI: 10.1093/aje/kwn073. PMID: 18420544.
- ↑ E. Cho, S.A. Smith-Warner, J. Ritz, P.A. van den Brandt i inni. Alcohol intake and colorectal cancer: a pooled analysis of 8 cohort studies. „Ann Intern Med”. 140 (8), s. 603–613, kwiecień 2004. PMID: 15096331.
- ↑ N.N. Baxter, J.E. Tepper, S.B. Durham, D.A. Rothenberger i inni. Increased risk of rectal cancer after prostate radiation: a population-based study. „Gastroenterology”. 128 (4), s. 819–824, kwiecień 2005. PMID: 15825064.
- ↑ B. Delhougne, C. Deneux, R. Abs, P. Chanson i inni. The prevalence of colonic polyps in acromegaly: a colonoscopic and pathological study in 103 patients. „J Clin Endocrinol Metab”. 80 (11), s. 3223–3226, listopad 1995. DOI: 10.1210/jcem.80.11.7593429. PMID: 7593429.
- ↑ A.G. Renehan, J. O’Connell, D. O’Halloran, F. Shanahan i inni. Acromegaly and colorectal cancer: a comprehensive review of epidemiology, biological mechanisms, and clinical implications. „Horm Metab Res”. 35 (11–12). s. 712–725. DOI: 10.1055/s-2004-814150. PMID: 14710350.
- ↑ a b c d F. Kastrinos, S. Syngal. Inherited colorectal cancer syndromes. „Cancer J”. 17 (6). s. 405–415. DOI: 10.1097/PPO.0b013e318237e408. PMID: 22157284.
- ↑ a b W.M. Grady. Genetic testing for high-risk colon cancer patients. „Gastroenterology”. 124 (6), s. 1574–1594, maj 2003. PMID: 12761718.
- ↑ a b c d e f E.M. Stoffel, F. Kastrinos. Familial colorectal cancer, beyond Lynch syndrome. „Clin Gastroenterol Hepatol”. 12 (7), s. 1059–1068, lipiec 2014. DOI: 10.1016/j.cgh.2013.08.015. PMID: 23962553.
- ↑ a b c J.M. Carethers, E.M. Stoffel. Lynch syndrome and Lynch syndrome mimics: The growing complex landscape of hereditary colon cancer. „World J Gastroenterol”. 21 (31), s. 9253–9261, sierpień 2015. DOI: 10.3748/wjg.v21.i31.9253. PMID: 26309352.
- ↑ K.E. Resnick, H. Hampel, R. Fishel, D.E. Cohn. Current and emerging trends in Lynch syndrome identification in women with endometrial cancer. „Gynecol Oncol”. 114 (1), s. 128–134, lipiec 2009. DOI: 10.1016/j.ygyno.2009.03.003. PMID: 19375789.
- ↑ A. de la Chapelle. The incidence of Lynch syndrome. „Fam Cancer”. 4 (3), s. 233–237, 2005. DOI: 10.1007/s10689-004-5811-3. PMID: 16136383.
- ↑ H. Hampel, W.L. Frankel, E. Martin, M. Arnold i inni. Screening for the Lynch syndrome (hereditary nonpolyposis colorectal cancer). „N Engl J Med”. 352 (18), s. 1851–1860, maj 2005. DOI: 10.1056/NEJMoa043146. PMID: 15872200.
- ↑ D.C. Chung, A.K. Rustgi. The hereditary nonpolyposis colorectal cancer syndrome: genetics and clinical implications. „Ann Intern Med”. 138 (7), s. 560–570, kwiecień 2003. PMID: 12667026.
- ↑ a b c V. Steinke, C. Engel, R. Büttner, H.K. Schackert i inni. Hereditary nonpolyposis colorectal cancer (HNPCC)/Lynch syndrome. „Dtsch Arztebl Int”. 110 (3), s. 32–38, styczeń 2013. DOI: 10.3238/arztebl.2013.0032. PMID: 23413378.
- ↑ G.L. Baiocchi, N. Portolani, W. Vermi, C. Baronchelli i inni. Lynch syndrome from a surgeon perspective: retrospective study of clinical impact of mismatch repair protein expression analysis in colorectal cancer patients less than 50 years old. „BMC Surg”. 14, s. 9, 2014. DOI: 10.1186/1471-2482-14-9. PMID: 24533633.
- ↑ a b M. Aarnio, R. Sankila, E. Pukkala, R. Salovaara i inni. Cancer risk in mutation carriers of DNA-mismatch-repair genes. „Int J Cancer”. 81 (2), s. 214–218, kwiecień 1999. PMID: 10188721.
- ↑ H.T. Lynch, T.C. Smyrk, P.M. Lynch, S.J. Lanspa i inni. Adenocarcinoma of the small bowel in lynch syndrome II. „Cancer”. 64 (10), s. 2178–2183, listopad 1989. PMID: 2804907.
- ↑ H.F. Vasen, J.T. Wijnen, F.H. Menko, J.H. Kleibeuker i inni. Cancer risk in families with hereditary nonpolyposis colorectal cancer diagnosed by mutation analysis. „Gastroenterology”. 110 (4), s. 1020–1027, kwiecień 1996. PMID: 8612988.
- ↑ Y. Parc, C. Boisson, G. Thomas, S. Olschwang. Cancer risk in 348 French MSH2 or MLH1 gene carriers. „J Med Genet”. 40 (3), s. 208–213, marzec 2003. PMID: 12624141.
- ↑ a b c d e f g J. Balmaña, F. Balaguer, A. Cervantes, D. Arnold. Familial risk-colorectal cancer: ESMO Clinical Practice Guidelines. „Ann Oncol”. 24 Suppl 6, s. vi73-80, październik 2013. DOI: 10.1093/annonc/mdt209. PMID: 23813931.
- ↑ Justyna Klusek, Stanisław Głuszek, Jolanta Klusek. Wybrane mutacje związane z dużym ryzykiem wystąpienia nowotworów jelita grubego. „Przegląd Gastroenterologiczny”, 2012.
- ↑ S. Shiovitz, W.K. Copeland, M.N. Passarelli, A.N. Burnett-Hartman i inni. Characterisation of familial colorectal cancer Type X, Lynch syndrome, and non-familial colorectal cancer. „Br J Cancer”. 111 (3), s. 598–602, lipiec 2014. DOI: 10.1038/bjc.2014.309. PMID: 24918813.
- ↑ F.G. Campos, M. Sulbaran, A.V. Safatle-Ribeiro, C.A. Martinez. Duodenal adenoma surveillance in patients with familial adenomatous polyposis. „World J Gastrointest Endosc”. 7 (10), s. 950–959, sierpień 2015. DOI: 10.4253/wjge.v7.i10.950. PMID: 26265988.
- ↑ a b c M.L. Leoz, S. Carballal, L. Moreira, T. Ocaña i inni. The genetic basis of familial adenomatous polyposis and its implications for clinical practice and risk management. „Appl Clin Genet”. 8, s. 95–107, 2015. DOI: 10.2147/TACG.S51484. PMID: 25931827.
- ↑ A.M. Jelsig, N. Qvist, K. Brusgaard, C.B. Nielsen i inni. Hamartomatous polyposis syndromes: a review. „Orphanet J Rare Dis”. 9, s. 101, 2014. DOI: 10.1186/1750-1172-9-101. PMID: 25022750.
- ↑ F.M. Giardiello, J.D. Brensinger, A.C. Tersmette, S.N. Goodman i inni. Very high risk of cancer in familial Peutz-Jeghers syndrome. „Gastroenterology”. 119 (6), s. 1447–1453, grudzień 2000. PMID: 11113065.
- ↑ a b N. Hearle, V. Schumacher, F.H. Menko, S. Olschwang i inni. Frequency and spectrum of cancers in the Peutz-Jeghers syndrome. „Clin Cancer Res”. 12 (10), s. 3209–3215, maj 2006. DOI: 10.1158/1078-0432.CCR-06-0083. PMID: 16707622.
- ↑ a b c F.G. Campos, M.N. Figueiredo, C.A. Martinez. Colorectal cancer risk in hamartomatous polyposis syndromes. „World J Gastrointest Surg”. 7 (3), s. 25–32, marzec 2015. DOI: 10.4240/wjgs.v7.i3.25. PMID: 25848489.
- ↑ a b M.G. Dunlop. Guidance on gastrointestinal surveillance for hereditary non-polyposis colorectal cancer, familial adenomatous polypolis, juvenile polyposis, and Peutz-Jeghers syndrome. „Gut”. 51 Suppl 5, s. V21-7, październik 2002. PMID: 12221036.
- ↑ E. Chow, F. Macrae. A review of juvenile polyposis syndrome. „J Gastroenterol Hepatol”. 20 (11), s. 1634–1640, listopad 2005. DOI: 10.1111/j.1440-1746.2005.03865.x. PMID: 16246179.
- ↑ N. Mishra, J. Hall. Identification of patients at risk for hereditary colorectal cancer. „Clin Colon Rectal Surg”. 25 (2), s. 67–82, czerwiec 2012. DOI: 10.1055/s-0032-1313777. PMID: 23730221.
- ↑ M.L. Poulsen, M.L. Bisgaard. MUTYH Associated Polyposis (MAP). „Curr Genomics”. 9 (6), s. 420–435, wrzesień 2008. DOI: 10.2174/138920208785699562. PMID: 19506731.
- ↑ a b c d A.T. Chan, E.L. Giovannucci. Primary prevention of colorectal cancer. „Gastroenterology”. 138 (6), s. 2029–2043.e10, czerwiec 2010. DOI: 10.1053/j.gastro.2010.01.057. PMID: 20420944.
- ↑ MayoClinic: Prevention. [dostęp 2015-10-27]. [zarchiwizowane z tego adresu (2015-10-27)].
- ↑ a b c d e Anna Kubiak, Witold Kycler, Maciej Trojanowski. Epidemiologia i profilaktyka raka jelita grubego w Polsce. „Probl Hig Epidemiol”, 2014.
- ↑ a b c d e f g h P.J. Tárraga López, J.S. Albero, J.A. Rodríguez-Montes. Primary and secondary prevention of colorectal cancer. „Clin Med Insights Gastroenterol”. 7, s. 33–46, 2014. DOI: 10.4137/CGast.S14039. PMID: 25093007.
- ↑ S.J. Winawer, A.G. Zauber, M.J. O’Brien, M.N. Ho i inni. Randomized comparison of surveillance intervals after colonoscopic removal of newly diagnosed adenomatous polyps. The National Polyp Study Workgroup. „N Engl J Med”. 328 (13), s. 901–906, kwiecień 1993. DOI: 10.1056/NEJM199304013281301. PMID: 8446136.
- ↑ F. Citarda, G. Tomaselli, R. Capocaccia, S. Barcherini i inni. Efficacy in standard clinical practice of colonoscopic polypectomy in reducing colorectal cancer incidence. „Gut”. 48 (6), s. 812–815, czerwiec 2001. PMID: 11358901.
- ↑ N.N. Baxter, M.A. Goldwasser, L.F. Paszat, R. Saskin i inni. Association of colonoscopy and death from colorectal cancer. „Ann Intern Med”. 150 (1), s. 1–8, styczeń 2009. PMID: 19075198.
- ↑ a b G.P. Young, E.L. Symonds, J.E. Allison, S.R. Cole i inni. Advances in Fecal Occult Blood Tests: the FIT revolution. „Dig Dis Sci”. 60 (3), s. 609–622, marzec 2015. DOI: 10.1007/s10620-014-3445-3. PMID: 25492500.
- ↑ P. Hewitson, P. Glasziou, L. Irwig, B. Towler i inni. Screening for colorectal cancer using the faecal occult blood test, Hemoccult. „Cochrane Database Syst Rev”, s. CD001216, 2007. DOI: 10.1002/14651858.CD001216.pub2. PMID: 17253456.
- ↑ a b c d F. Stracci, M. Zorzi, G. Grazzini. Colorectal cancer screening: tests, strategies, and perspectives. „Front Public Health”. 2, s. 210, 2014. DOI: 10.3389/fpubh.2014.00210. PMID: 25386553.
- ↑ E. Flossmann, P.M. Rothwell. Effect of aspirin on long-term risk of colorectal cancer: consistent evidence from randomised and observational studies. „Lancet”. 369 (9573), s. 1603–1613, maj 2007. DOI: 10.1016/S0140-6736(07)60747-8. PMID: 17499602.
- ↑ T.K. Asano, R.S. McLeod. Non steroidal anti-inflammatory drugs (NSAID) and Aspirin for preventing colorectal adenomas and carcinomas. „Cochrane Database Syst Rev”, s. CD004079, 2004. DOI: 10.1002/14651858.CD004079.pub2. PMID: 15106236.
- ↑ Y.P. Wang, Q. Wang, T. Gan, T. Pan i inni. Non-steroidal anti-inflammatory agents for chemoprevention of colorectal polyps: a meta-analysis. „Zhonghua Nei Ke Za Zhi”. 43 (1), s. 10–12, styczeń 2004. PMID: 14990012.
- ↑ A.T. Chan, E.L. Giovannucci, J.A. Meyerhardt, E.S. Schernhammer i inni. Aspirin dose and duration of use and risk of colorectal cancer in men. „Gastroenterology”. 134 (1), s. 21–28, styczeń 2008. DOI: 10.1053/j.gastro.2007.09.035. PMID: 18005960.
- ↑ N. Arber, C.J. Eagle, J. Spicak, I. Rácz i inni. Celecoxib for the prevention of colorectal adenomatous polyps. „N Engl J Med”. 355 (9), s. 885–895, sierpień 2006. DOI: 10.1056/NEJMoa061652. PMID: 16943401.
- ↑ M.M. Bertagnolli, C.J. Eagle, A.G. Zauber, M. Redston i inni. Celecoxib for the prevention of sporadic colorectal adenomas. „N Engl J Med”. 355 (9), s. 873–884, sierpień 2006. DOI: 10.1056/NEJMoa061355. PMID: 16943400.
- ↑ J.A. Baron, R.S. Sandler, R.S. Bresalier, H. Quan i inni. A randomized trial of rofecoxib for the chemoprevention of colorectal adenomas. „Gastroenterology”. 131 (6), s. 1674–1682, grudzień 2006. DOI: 10.1053/j.gastro.2006.08.079. PMID: 17087947.
- ↑ S.D. Solomon, M.A. Pfeffer, J.J. McMurray, R. Fowler i inni. Effect of celecoxib on cardiovascular events and blood pressure in two trials for the prevention of colorectal adenomas. „Circulation”. 114 (10), s. 1028–1035, wrzesień 2006. DOI: 10.1161/CIRCULATIONAHA.106.636746. PMID: 16943394.
- ↑ R.S. Bresalier, R.S. Sandler, H. Quan, J.A. Bolognese i inni. Cardiovascular events associated with rofecoxib in a colorectal adenoma chemoprevention trial. „N Engl J Med”. 352 (11), s. 1092–1102, marzec 2005. DOI: 10.1056/NEJMoa050493. PMID: 15713943.
- ↑ F.M. Giardiello, S.R. Hamilton, A.J. Krush, S. Piantadosi i inni. Treatment of colonic and rectal adenomas with sulindac in familial adenomatous polyposis. „N Engl J Med”. 328 (18), s. 1313–1316, maj 1993. DOI: 10.1056/NEJM199305063281805. PMID: 8385741.
- ↑ D. Labayle, D. Fischer, P. Vielh, F. Drouhin i inni. Sulindac causes regression of rectal polyps in familial adenomatous polyposis. „Gastroenterology”. 101 (3), s. 635–639, wrzesień 1991. PMID: 1650315.
- ↑ S. Bonovas, K. Filioussi, C.S. Flordellis, N.M. Sitaras. Statins and the risk of colorectal cancer: a meta-analysis of 18 studies involving more than 1.5 million patients. „J Clin Oncol”. 25 (23), s. 3462–3468, sierpień 2007. DOI: 10.1200/JCO.2007.10.8936. PMID: 17687150.
- ↑ WHO Disease and injury country estimates. World Health Organization, 2009. [dostęp 2015-04-15].
- ↑ S. Liu, R. Zheng, M. Zhang, S. Zhang i inni. Incidence and mortality of colorectal cancer in China, 2011. „Chin J Cancer Res”. 27 (1), s. 22–28, luty 2015. DOI: 10.3978/j.issn.1000-9604.2015.02.01. PMID: 25717222.
- ↑ a b c d e f Krajowy Rejestr Nowotworów: Nowotwory złośliwe jelita grubego (C18-21). [dostęp 2015-10-26]. [zarchiwizowane z tego adresu (2015-10-26)].
- ↑ a b Van Cutsem i in. 2014 ↓, s. 1.
- ↑ J. Ferlay, E. Steliarova-Foucher, J. Lortet-Tieulent, S. Rosso i inni. Cancer incidence and mortality patterns in Europe: estimates for 40 countries in 2012. „Eur J Cancer”. 49 (6), s. 1374–1403, kwiecień 2013. DOI: 10.1016/j.ejca.2012.12.027. PMID: 23485231.
- ↑ a b c R. Siegel, C. Desantis, A. Jemal. Colorectal cancer statistics, 2014. „CA Cancer J Clin”. 64 (2). s. 104–117. DOI: 10.3322/caac.21220. PMID: 24639052.
- ↑ a b c F. Amersi, M. Agustin, C.Y. Ko. Colorectal cancer: epidemiology, risk factors, and health services. „Clin Colon Rectal Surg”. 18 (3), s. 133–140, sierpień 2005. DOI: 10.1055/s-2005-916274. PMID: 20011296.
- ↑ a b c G. Murphy, S.S. Devesa, A.J. Cross, P.D. Inskip i inni. Sex disparities in colorectal cancer incidence by anatomic subsite, race and age. „Int J Cancer”. 128 (7), s. 1668–1675, kwiecień 2011. DOI: 10.1002/ijc.25481. PMID: 20503269.
- ↑ a b R.J. Wong. Marked variations in proximal colon cancer survival by race/ethnicity within the United States. „J Clin Gastroenterol”. 44 (9), s. 625–630, październik 2010. DOI: 10.1097/MCG.0b013e3181c64a7a. PMID: 19996985.
- ↑ Piotr Potemski. Epidemiologia, badania przesiewowe i klasyfikacja zaawansowania klinicznego raka jelita grubego. „Onkologia w Praktyce Klinicznej”, 2010.
- ↑ Jan Korniluk, Gabriel Wcisło, Paweł Nurzyński, Rafał Stec i inni. Epidemiologia raka jelita grubego. „Współczesna Onkologia”, 2006.
- ↑ Hamilton i Aaltonen 2000 ↓, s. 104.
- ↑ a b Anna Nasierowska-Guttmejer, Barbara Górnicka: Zalecenia do diagnostyki histopatologicznej nowotworów. Warszawa: Centrum Onkologii, Oddział Gliwice; Polskie Towarzystwo Patologów, 2013, s. 101. ISBN 978-83-909137-1-1.
- ↑ a b c d e f g h i j k l m n o p M. Fleming, S. Ravula, S.F. Tatishchev, H.L. Wang. Colorectal carcinoma: Pathologic aspects. „J Gastrointest Oncol”. 3 (3), s. 153–173, wrzesień 2012. DOI: 10.3978/j.issn.2078-6891.2012.030. PMID: 22943008.
- ↑ a b Podolsky i in. 2015 ↓, s. 1570.
- ↑ Cassidy, Johnston i Van Cutsem 2006 ↓, s. 106.
- ↑ Kumar, Cotran i Robins 2005 ↓, s. 666.
- ↑ William B. Coleman, Gregory J. Gregory J.: The Molecular Basis of Human Cancer. Springer Science & Business Media, 2001, s. 12. ISBN 978-1-59259-125-1.
- ↑ a b Hamilton i Aaltonen 2000 ↓, s. 106.
- ↑ L. Cheng, C. Eng, L.Z. Nieman, A.S. Kapadia i inni. Trends in colorectal cancer incidence by anatomic site and disease stage in the United States from 1976 to 2005. „Am J Clin Oncol”. 34 (6), s. 573–580, grudzień 2011. DOI: 10.1097/COC.0b013e3181fe41ed. PMID: 21217399.
- ↑ a b c M.A. Jafarabadi, S.M. Mohammadi, E. Hajizadeh, A. Kazemnejad i inni. Does the prognosis of colorectal cancer vary with tumor site?. „Gastroenterol Hepatol Bed Bench”. 4 (4), s. 199–209, 2011. PMID: 24834183.
- ↑ N. Savas, U. Dagli, S. Akbulut, O. Yuksel i inni. Colorectal cancer localization in young patients: should we expand the screening program?. „Dig Dis Sci”. 52 (3), s. 798–802, marzec 2007. DOI: 10.1007/s10620-006-9432-6. PMID: 17245629.
- ↑ R.A. Meguid, M.B. Slidell, C.L. Wolfgang, D.C. Chang i inni. Is there a difference in survival between right- versus left-sided colon cancers?. „Ann Surg Oncol”. 15 (9), s. 2388–2394, wrzesień 2008. DOI: 10.1245/s10434-008-0015-y. PMID: 18622647.
- ↑ A.K. Lam, S.S. Chan, M. Leung. Synchronous colorectal cancer: clinical, pathological and molecular implications. „World J Gastroenterol”. 20 (22), s. 6815–6820, czerwiec 2014. DOI: 10.3748/wjg.v20.i22.6815. PMID: 24944471.
- ↑ a b c Hamilton i Aaltonen 2000 ↓, s. 108.
- ↑ Kumar, Cotran i Robins 2005 ↓, s. 669.
- ↑ a b Kelsen 2008 ↓, s. 555.
- ↑ Kelsen 2008 ↓, s. 18–19.
- ↑ Price i Sikora 2014 ↓, s. 313.
- ↑ a b Odze i Goldblum 2009 ↓, s. 606.
- ↑ Hamilton i Aaltonen 2000 ↓, s. 110–111.
- ↑ a b c Odze i Goldblum 2009 ↓, s. 607.
- ↑ a b c d e Hamilton i Aaltonen 2000 ↓, s. 109.
- ↑ a b c d e Odze i Goldblum 2009 ↓, s. 609.
- ↑ a b c Hamilton i Aaltonen 2000 ↓, s. 110.
- ↑ a b Kelsen 2008 ↓, s. 558.
- ↑ Odze i Goldblum 2009 ↓, s. 610.
- ↑ Odze i Goldblum 2009 ↓, s. 611.
- ↑ a b c d e f N.S. Goldstein. Serrated pathway and APC (conventional)-type colorectal polyps: molecular-morphologic correlations, genetic pathways, and implications for classification. „Am J Clin Pathol”. 125 (1), s. 146–153, styczeń 2006. PMID: 16483003.
- ↑ a b c d Janina Orłowska, Mirosław Kiedrowski. Gruczolaki ząbkowane i polipowatość hyperplastyczna, a rak jelita grubego. „Postępy Nauk Medycznych”, 2009.
- ↑ a b c Hamilton i Aaltonen 2000 ↓, s. 111.
- ↑ a b c d J. Munding, A. Tannapfel. Epidemiology of Colorectal Adenomas and Histopathological Assessment of Endoscopic Specimens in the Colorectum. „Viszeralmedizin”. 30 (1), s. 10–16, luty 2014. DOI: 10.1159/000357744. PMID: 26288577.
- ↑ a b c Janina Orłowska, Jacek Pachlewski: Zmiany ząbkowane i polipowatość ząbkowana (hiperplastyczna) a rak jelita grubego. [dostęp 2015-10-31]. [zarchiwizowane z tego adresu (2015-10-31)].
- ↑ a b c D.K. Rex, D.J. Ahnen, J.A. Baron, K.P. Batts i inni. Serrated lesions of the colorectum: review and recommendations from an expert panel. „Am J Gastroenterol”. 107 (9), s. 1315–1329; quiz 1314, 1330, wrzesień 2012. DOI: 10.1038/ajg.2012.161. PMID: 22710576.
- ↑ a b c d Mróz Andrzej. Diagnostyka różnicowa zmian przedrakowych: polipy i zespoły polipowatości jelita (zespoły polipów uwarunkowane dziedzicznie). „Pol J Pathol”, 2014.
- ↑ Hamilton i Aaltonen 2000 ↓, s. 112.
- ↑ a b V.P. Bauer, H.T. Papaconstantinou. Management of serrated adenomas and hyperplastic polyps. „Clin Colon Rectal Surg”. 21 (4), s. 273–279, listopad 2008. DOI: 10.1055/s-0028-1089942. PMID: 20011438.
- ↑ Levin i in. 2005 ↓, s. 30–31.
- ↑ a b J.J. Centelles. General aspects of colorectal cancer. „ISRN Oncol”. 2012, s. 139268, 2012. DOI: 10.5402/2012/139268. PMID: 23209942.
- ↑ a b Levin i in. 2005 ↓, s. 30.
- ↑ Levin i in. 2005 ↓, s. 31.
- ↑ a b c d Karen E. Kim: Early Detection and Prevention of Colorectal Cancer. SLACK Incorporated, 2009, s. 20–21. ISBN 978-1-55642-837-1.
- ↑ a b Leonard B. Saltz: Colorectal Cancer: Multimodality Management. Springer Science & Business Media, 2002, s. 82. ISBN 978-1-59259-160-2.
- ↑ a b N. Shussman, S.D. Wexner. Colorectal polyps and polyposis syndromes. „Gastroenterol Rep (Oxf)”. 2 (1), s. 1–15, luty 2014. DOI: 10.1093/gastro/got041. PMID: 24760231.
- ↑ a b D.L. Worthley, V.L. Whitehall, K.J. Spring, B.A. Leggett. Colorectal carcinogenesis: road maps to cancer. „World J Gastroenterol”. 13 (28), s. 3784–3791, lipiec 2007. PMID: 17657831.
- ↑ a b c d e f g h i H. Raskov, H.C. Pommergaard, J. Burcharth, J. Rosenberg. Colorectal carcinogenesis-update and perspectives. „World J Gastroenterol”. 20 (48), s. 18151–18164, grudzień 2014. DOI: 10.3748/wjg.v20.i48.18151. PMID: 25561783.
- ↑ a b c A.M. Joosen, G.G. Kuhnle, S.M. Aspinall, T.M. Barrow i inni. Effect of processed and red meat on endogenous nitrosation and DNA damage. „Carcinogenesis”. 30 (8), s. 1402–1407, sierpień 2009. DOI: 10.1093/carcin/bgp130. PMID: 19498009.
- ↑ a b c d e f g R.L. Santarelli, F. Pierre, D.E. Corpet. Processed meat and colorectal cancer: a review of epidemiologic and experimental evidence. „Nutr Cancer”. 60 (2), s. 131–144, 2008. DOI: 10.1080/01635580701684872. PMID: 18444144.
- ↑ W. Lijinsky. N-Nitroso compounds in the diet. „Mutat Res”. 443 (1–2), s. 129–138, lipiec 1999. PMID: 10415436.
- ↑ C.T. Dellavalle, Q. Xiao, G. Yang, X.O. Shu i inni. Dietary nitrate and nitrite intake and risk of colorectal cancer in the Shanghai Women’s Health Study. „Int J Cancer”. 134 (12), s. 2917–2926, czerwiec 2014. DOI: 10.1002/ijc.28612. PMID: 24242755.
- ↑ S.A. Bingham, R. Hughes, A.J. Cross. Effect of white versus red meat on endogenous N-nitrosation in the human colon and further evidence of a dose response. „J Nutr”. 132 (11 Suppl), s. 3522S-3525S, listopad 2002. PMID: 12421881.
- ↑ a b A.J. Cross, J.R. Pollock, S.A. Bingham. Haem, not protein or inorganic iron, is responsible for endogenous intestinal N-nitrosation arising from red meat. „Cancer Res”. 63 (10), s. 2358–2360, maj 2003. PMID: 12750250.
- ↑ A.L. Sesink, D.S. Termont, J.H. Kleibeuker, R. Van der Meer. Red meat and colon cancer: the cytotoxic and hyperproliferative effects of dietary heme. „Cancer Res”. 59 (22), s. 5704–5709, listopad 1999. PMID: 10582688.
- ↑ T. Sawa, T. Akaike, K. Kida, Y. Fukushima i inni. Lipid peroxyl radicals from oxidized oils and heme-iron: implication of a high-fat diet in colon carcinogenesis. „Cancer Epidemiol Biomarkers Prev”. 7 (11), s. 1007–1012, listopad 1998. PMID: 9829709.
- ↑ S. Nowell, B. Coles, R. Sinha, S. MacLeod i inni. Analysis of total meat intake and exposure to individual heterocyclic amines in a case-control study of colorectal cancer: contribution of metabolic variation to risk. „Mutat Res”. 506–507, s. 175–185, wrzesień 2002. PMID: 12351157.
- ↑ M. Scarpa, I. Castagliuolo, C. Castoro, A. Pozza i inni. Inflammatory colonic carcinogenesis: a review on pathogenesis and immunosurveillance mechanisms in ulcerative colitis. „World J Gastroenterol”. 20 (22), s. 6774–6785, czerwiec 2014. DOI: 10.3748/wjg.v20.i22.6774. PMID: 24944468.
- ↑ a b c J.K. Triantafillidis, G. Nasioulas, P.A. Kosmidis. Colorectal cancer and inflammatory bowel disease: epidemiology, risk factors, mechanisms of carcinogenesis and prevention strategies. „Anticancer Res”. 29 (7), s. 2727–2737, lipiec 2009. PMID: 19596953.
- ↑ B. Yan, H. Wang, Z.N. Rabbani, Y. Zhao i inni. Tumor necrosis factor-alpha is a potent endogenous mutagen that promotes cellular transformation. „Cancer Res”. 66 (24), s. 11565–11570, grudzień 2006. DOI: 10.1158/0008-5472.CAN-06-2540. PMID: 17178846.
- ↑ A.J. Schottelius, H. Dinter. Cytokines, NF-kappaB, microenvironment, intestinal inflammation and cancer. „Cancer Treat Res”. 130, s. 67–87, 2006. PMID: 16610703.
- ↑ M.M. Derry, K. Raina, C. Agarwal, R. Agarwal. Identifying molecular targets of lifestyle modifications in colon cancer prevention. „Front Oncol”. 3, s. 119, 2013. DOI: 10.3389/fonc.2013.00119. PMID: 23675573.
- ↑ a b c d e f g h i j k F. Arvelo, F. Sojo, C. Cotte. Biology of colorectal cancer. „Ecancermedicalscience”. 9, s. 520, 2015. DOI: 10.3332/ecancer.2015.520. PMID: 25932044.
- ↑ B. Li, X.Y. Shi, D.X. Liao, B.R. Cao i inni. Advanced colorectal adenoma related gene expression signature may predict prognostic for colorectal cancer patients with adenoma-carcinoma sequence. „Int J Clin Exp Med”. 8 (4), s. 4883–4898, 2015. PMID: 26131062.
- ↑ a b c d e f g h i j k l m n o T. Armaghany, J.D. Wilson, Q. Chu, G. Mills. Genetic alterations in colorectal cancer. „Gastrointest Cancer Res”. 5 (1), s. 19–27, styczeń 2012. PMID: 22574233.
- ↑ a b c L. Yamane, C. Scapulatempo-Neto, R.M. Reis, D.P. Guimarães. Serrated pathway in colorectal carcinogenesis. „World J Gastroenterol”. 20 (10), s. 2634–2640, marzec 2014. DOI: 10.3748/wjg.v20.i10.2634. PMID: 24627599.
- ↑ E. Vilar, S.B. Gruber. Microsatellite instability in colorectal cancer-the stable evidence. „Nat Rev Clin Oncol”. 7 (3), s. 153–162, marzec 2010. DOI: 10.1038/nrclinonc.2009.237. PMID: 20142816.
- ↑ D. Kedrin, M.K. Gala. Genetics of the serrated pathway to colorectal cancer. „Clin Transl Gastroenterol”. 6, s. e84, 2015. DOI: 10.1038/ctg.2015.12. PMID: 25856207.
- ↑ Ł. Szylberg, M. Janiczek, A. Popiel, A. Marszałek. Serrated polyps and their alternative pathway to the colorectal cancer: a systematic review. „Gastroenterol Res Pract”. 2015, s. 573814, 2015. DOI: 10.1155/2015/573814. PMID: 25945086.
- ↑ P. Alberici, R. Fodde. The role of the APC tumor suppressor in chromosomal instability. „Genome Dyn”. 1, s. 149–170, 2006. DOI: 10.1159/000092506. PMID: 18724059.
- ↑ R.H. Giles, J.H. van Es, H. Clevers. Caught up in a Wnt storm: Wnt signaling in cancer. „Biochim Biophys Acta”. 1653 (1), s. 1–24, czerwiec 2003. PMID: 12781368.
- ↑ T. Takayama, M. Ohi, T. Hayashi, K. Miyanishi i inni. Analysis of K-ras, APC, and beta-catenin in aberrant crypt foci in sporadic adenoma, cancer, and familial adenomatous polyposis. „Gastroenterology”. 121 (3), s. 599–611, wrzesień 2001. PMID: 11522744.
- ↑ K. Aoki, M.M. Taketo. Adenomatous polyposis coli (APC): a multi-functional tumor suppressor gene. „J Cell Sci”. 120 (Pt 19), s. 3327–3335, październik 2007. DOI: 10.1242/jcs.03485. PMID: 17881494.
- ↑ a b c X.L. Li, J. Zhou, Z.R. Chen, W.J. Chng. P53 mutations in colorectal cancer – molecular pathogenesis and pharmacological reactivation. „World J Gastroenterol”. 21 (1), s. 84–93, styczeń 2015. DOI: 10.3748/wjg.v21.i1.84. PMID: 25574081.
- ↑ A. Russo, V. Bazan, B. Iacopetta, D. Kerr i inni. The TP53 colorectal cancer international collaborative study on the prognostic and predictive significance of p53 mutation: influence of tumor site, type of mutation, and adjuvant treatment. „J Clin Oncol”. 23 (30), s. 7518–7528, październik 2005. DOI: 10.1200/JCO.2005.00.471. PMID: 16172461.
- ↑ B. Vogelstein, E.R. Fearon, S.R. Hamilton, S.E. Kern i inni. Genetic alterations during colorectal-tumor development. „N Engl J Med”. 319 (9), s. 525–532, wrzesień 1988. DOI: 10.1056/NEJM198809013190901. PMID: 2841597.
- ↑ a b c M. Duman-Scheel. Deleted in Colorectal Cancer (DCC) pathfinding: axon guidance gene finally turned tumor suppressor. „Curr Drug Targets”. 13 (11), s. 1445–1453, październik 2012. PMID: 22876889.
- ↑ C. Forcet, X. Ye, L. Granger, V. Corset i inni. The dependence receptor DCC (deleted in colorectal cancer) defines an alternative mechanism for caspase activation. „Proc Natl Acad Sci U S A”. 98 (6), s. 3416–3421, marzec 2001. DOI: 10.1073/pnas.051378298. PMID: 11248093.
- ↑ a b c A.M. Krasinskas. EGFR Signaling in Colorectal Carcinoma. „Patholog Res Int”. 2011, s. 932932, 2011. DOI: 10.4061/2011/932932. PMID: 21403829.
- ↑ a b J.P. Spano, R. Fagard, J.C. Soria, O. Rixe i inni. Epidermal growth factor receptor signaling in colorectal cancer: preclinical data and therapeutic perspectives. „Ann Oncol”. 16 (2), s. 189–194, luty 2005. DOI: 10.1093/annonc/mdi057. PMID: 15668269.
- ↑ a b c d C. Tan, X. Du. KRAS mutation testing in metastatic colorectal cancer. „World J Gastroenterol”. 18 (37), s. 5171–5180, październik 2012. DOI: 10.3748/wjg.v18.i37.5171. PMID: 23066310.
- ↑ a b C.S. Karapetis, S. Khambata-Ford, D.J. Jonker, C.J. O’Callaghan i inni. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. „N Engl J Med”. 359 (17), s. 1757–1765, październik 2008. DOI: 10.1056/NEJMoa0804385. PMID: 18946061.
- ↑ a b R.G. Amado, M. Wolf, M. Peeters, E. Van Cutsem i inni. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. „J Clin Oncol”. 26 (10), s. 1626–1634, kwiecień 2008. DOI: 10.1200/JCO.2007.14.7116. PMID: 18316791.
- ↑ a b c M. Peeters, T.J. Price, A. Cervantes, A.F. Sobrero i inni. Randomized phase III study of panitumumab with fluorouracil, leucovorin, and irinotecan (FOLFIRI) compared with FOLFIRI alone as second-line treatment in patients with metastatic colorectal cancer. „J Clin Oncol”. 28 (31), s. 4706–4713, listopad 2010. DOI: 10.1200/JCO.2009.27.6055. PMID: 20921462.
- ↑ a b E. Van Cutsem, C.H. Köhne, I. Láng, G. Folprecht i inni. Cetuximab plus irinotecan, fluorouracil, and leucovorin as first-line treatment for metastatic colorectal cancer: updated analysis of overall survival according to tumor KRAS and BRAF mutation status. „J Clin Oncol”. 29 (15), s. 2011–2019, maj 2011. DOI: 10.1200/JCO.2010.33.5091. PMID: 21502544.
- ↑ J. Colicelli. Human RAS superfamily proteins and related GTPases. „Sci STKE”. 2004 (250), s. RE13, wrzesień 2004. DOI: 10.1126/stke.2502004re13. PMID: 15367757.
- ↑ D. Vigil, J. Cherfils, K.L. Rossman, C.J. Der. Ras superfamily GEFs and GAPs: validated and tractable targets for cancer therapy?. „Nat Rev Cancer”. 10 (12), s. 842–857, grudzień 2010. DOI: 10.1038/nrc2960. PMID: 21102635.
- ↑ X. Liu, M. Jakubowski, J.L. Hunt. KRAS gene mutation in colorectal cancer is correlated with increased proliferation and spontaneous apoptosis. „Am J Clin Pathol”. 135 (2), s. 245–252, luty 2011. DOI: 10.1309/AJCP7FO2VAXIVSTP. PMID: 21228365.
- ↑ D. Barras. BRAF Mutation in Colorectal Cancer: An Update. „Biomark Cancer”. 7 (Suppl 1), s. 9–12, 2015. DOI: 10.4137/BIC.S25248. PMID: 26396549.
- ↑ F.A. Sinicrope, Q. Shi, T.C. Smyrk, S.N. Thibodeau i inni. Molecular markers identify subtypes of stage III colon cancer associated with patient outcomes. „Gastroenterology”. 148 (1), s. 88–99, styczeń 2015. DOI: 10.1053/j.gastro.2014.09.041. PMID: 25305506.
- ↑ B. Tran, S. Kopetz, J. Tie, P. Gibbs i inni. Impact of BRAF mutation and microsatellite instability on the pattern of metastatic spread and prognosis in metastatic colorectal cancer. „Cancer”. 117 (20), s. 4623–4632, październik 2011. DOI: 10.1002/cncr.26086. PMID: 21456008.
- ↑ a b c C.R. Boland, A. Goel. Microsatellite instability in colorectal cancer. „Gastroenterology”. 138 (6), s. 2073–2087.e3, czerwiec 2010. DOI: 10.1053/j.gastro.2009.12.064. PMID: 20420947.
- ↑ P. Peltomäki. Deficient DNA mismatch repair: a common etiologic factor for colon cancer. „Hum Mol Genet”. 10 (7), s. 735–740, kwiecień 2001. PMID: 11257106.
- ↑ R. Jover, A. Payá, C. Alenda, M.J. Poveda i inni. Defective mismatch-repair colorectal cancer: clinicopathologic characteristics and usefulness of immunohistochemical analysis for diagnosis. „Am J Clin Pathol”. 122 (3), s. 389–394, wrzesień 2004. DOI: 10.1309/V9PG-K2Y2-60VF-VULR. PMID: 15362369.
- ↑ M. Pinheiro, T. Ahlquist, S.A. Danielsen, G.E. Lind i inni. Colorectal carcinomas with microsatellite instability display a different pattern of target gene mutations according to large bowel site of origin. „BMC Cancer”. 10, s. 587, 2010. DOI: 10.1186/1471-2407-10-587. PMID: 20979647.
- ↑ L.M. Ellis, Y. Takahashi, W. Liu, R.M. Shaheen. Vascular endothelial growth factor in human colon cancer: biology and therapeutic implications. „Oncologist”. 5 Suppl 1, s. 11–15, 2000. PMID: 10804085.
- ↑ a b R. Bendardaf, A. Buhmeida, M. Hilska, M. Laato i inni. VEGF-1 expression in colorectal cancer is associated with disease localization, stage, and long-term disease-specific survival. „Anticancer Res”. 28 (6B). s. 3865–3870. PMID: 19192642.
- ↑ a b c D. Cao, M. Hou, Y.S. Guan, M. Jiang i inni. Expression of HIF-1alpha and VEGF in colorectal cancer: association with clinical outcomes and prognostic implications. „BMC Cancer”. 9, s. 432, 2009. DOI: 10.1186/1471-2407-9-432. PMID: 20003271.
- ↑ M. Mathonnet, A. Perraud, N. Christou, H. Akil i inni. Hallmarks in colorectal cancer: angiogenesis and cancer stem-like cells. „World J Gastroenterol”. 20 (15), s. 4189–4196, kwiecień 2014. DOI: 10.3748/wjg.v20.i15.4189. PMID: 24764657.
- ↑ Dermot R. Fitzgibbon, John D. Loeser: Cancer Pain. Lippincott Williams & Wilkins, 2012, s. 50. ISBN 978-1-4511-5279-1.
- ↑ S.J. Winawer. Natural history of colorectal cancer. „Am J Med”. 106 (1A), s. 3S-6S; discussion 50S-51S, styczeń 1999. PMID: 10089106.
- ↑ S.J. Stryker, B.G. Wolff, C.E. Culp, S.D. Libbe i inni. Natural history of untreated colonic polyps. „Gastroenterology”. 93 (5), s. 1009–1013, listopad 1987. PMID: 3653628.
- ↑ W.S. Atkin, B.P. Saunders. Surveillance guidelines after removal of colorectal adenomatous polyps. „Gut”. 51 Suppl 5, s. V6-9, październik 2002. PMID: 12221031.
- ↑ a b Podolsky i in. 2015 ↓, s. 1571.
- ↑ T. Matsui, T. Yao, A. Iwashita. Natural history of early colorectal cancer. „World J Surg”. 24 (9), s. 1022–1028, wrzesień 2000. PMID: 11036277.
- ↑ a b Bailey i in. 2012 ↓, s. 238.
- ↑ R.G. Landmann, M.R. Weiser. Surgical management of locally advanced and locally recurrent colon cancer. „Clin Colon Rectal Surg”. 18 (3), s. 182–189, sierpień 2005. DOI: 10.1055/s-2005-916279. PMID: 20011301.
- ↑ a b Robin K.S. Phillips, Sue Clark: Colorectal Surgery: Companion to Specialist Surgical Practice. Elsevier Health Sciences, 2013. ISBN 978-0-7020-4973-6.
- ↑ a b Odze i Goldblum 2009 ↓, s. 617.
- ↑ DeVita, Lawrence i Rosenberg 2008 ↓, s. 1245.
- ↑ C. Liebig, G. Ayala, J.A. Wilks, D.H. Berger i inni. Perineural invasion in cancer: a review of the literature. „Cancer”. 115 (15), s. 3379–3391, sierpień 2009. DOI: 10.1002/cncr.24396. PMID: 19484787.
- ↑ N. Knijn, S.C. Mogk, S. Teerenstra, F. Simmer i inni. Perineural Invasion Is a Strong Prognostic Factor in Colorectal Cancer: A Systematic Review. „Am J Surg Pathol”, wrzesień 2015. DOI: 10.1097/PAS.0000000000000518. PMID: 26426380.
- ↑ a b c M. Qiu, J. Hu, D. Yang, D.P. Cosgrove i inni. Pattern of distant metastases in colorectal cancer: a SEER based study. „Oncotarget”, październik 2015. DOI: 10.18632/oncotarget.6130. PMID: 26484417.
- ↑ P. Gervaz, P. Bucher, I. Neyroud-Caspar, C. Soravia i inni. Proximal location of colon cancer is a risk factor for development of metachronous colorectal cancer: a population-based study. „Dis Colon Rectum”. 48 (2), s. 227–232, luty 2005. DOI: 10.1007/s10350-004-0805-7. PMID: 15711864.
- ↑ J. Kaplan, A. Strongin, D.G. Adler, A.A. Siddiqui. Enteral stents for the management of malignant colorectal obstruction. „World J Gastroenterol”. 20 (37), s. 13239–13245, październik 2014. DOI: 10.3748/wjg.v20.i37.13239. PMID: 25309061.
- ↑ S.P. Hong, T.I. Kim. Colorectal stenting: an advanced approach to malignant colorectal obstruction. „World J Gastroenterol”. 20 (43), s. 16020–16028, listopad 2014. DOI: 10.3748/wjg.v20.i43.16020. PMID: 25473154.
- ↑ a b c M. Ogawa, M. Watanabe, K. Eto, T. Omachi i inni. Clinicopathological features of perforated colorectal cancer. „Anticancer Res”. 29 (5), s. 1681–1684, maj 2009. PMID: 19443386.
- ↑ a b Alfred E. Chang, Patricia A. Ganz, Daniel F. Hayes, Timothy Kinsella, Harvey I. Pass, Joan H. Schiller, Richard M. Stone, Victor Strecher: Oncology: An Evidence-Based Approach. Springer Science & Business Media, 2007, s. 709. ISBN 978-0-387-31056-5.
- ↑ a b Ho 2012 ↓, s. 80–81.
- ↑ Jeffrey Norton, Philip S. Barie, Ralph R. Bollinger, Alfred E. Chang, Stephen Lowry, Sean J. Mulvihill, Harvey I. Pass, Robert W. Thompson: Surgery: Basic Science and Clinical Evidence. Springer Science & Business Media, 2009, s. 1069. ISBN 978-0-387-68113-9.
- ↑ a b c d e f X.F. Yang, K. Pan. Diagnosis and management of acute complications in patients with colon cancer: bleeding, obstruction, and perforation. „Chin J Cancer Res”. 26 (3), s. 331–340, czerwiec 2014. DOI: 10.3978/j.issn.1000-9604.2014.06.11. PMID: 25035661.
- ↑ a b c d e f g h i M. Świderska, B. Choromańska, E. Dąbrowska, E. Konarzewska-Duchnowska i inni. The diagnostics of colorectal cancer. „Contemp Oncol (Pozn)”. 18 (1), s. 1–6, 2014. DOI: 10.5114/wo.2013.39995. PMID: 24876814.
- ↑ a b c d e f D.A. Fisher, A.K. Shergill, D.S. Early, R.D. Acosta i inni. Role of endoscopy in the staging and management of colorectal cancer. „Gastrointest Endosc”. 78 (1), s. 8–12, lipiec 2013. DOI: 10.1016/j.gie.2013.04.163. PMID: 23664162.
- ↑ Willett 2001 ↓, s. 82.
- ↑ a b R.J. Loffeld, M. Flens, G. Fransen, F.C. den Boer i inni. The localisation of cancer in the sigmoid, rectum or rectosigmoid junction using endoscopy or radiology-What is the most accurate method?. „J Gastrointest Oncol”. 5 (6), s. 469–473, grudzień 2014. DOI: 10.3978/j.issn.2078-6891.2014.087. PMID: 25436127.
- ↑ a b c S.E. Araujo, P.R. Alves, A. Habr-Gama. Role of colonoscopy in colorectal cancer. „Rev Hosp Clin Fac Med Sao Paulo”. 56 (1). s. 25–35. PMID: 11378680.
- ↑ Jerzy Gil, Stanisław Wojtuń. Diagnostyka endoskopowa raka jelita grubego. „Współczesna Onkologia”. 10, 2006.
- ↑ a b c d A. Laghi. Computed tomography colonography in 2014: an update on technique and indications. „World J Gastroenterol”. 20 (45), s. 16858–16867, grudzień 2014. DOI: 10.3748/wjg.v20.i45.16858. PMID: 25492999.
- ↑ a b S. Bipat, A.S. Glas, F.J. Slors, A.H. Zwinderman i inni. Rectal cancer: local staging and assessment of lymph node involvement with endoluminal US, CT, and MR imaging-a meta-analysis. „Radiology”. 232 (3), s. 773–783, wrzesień 2004. DOI: 10.1148/radiol.2323031368. PMID: 15273331.
- ↑ S.R. Puli, M.L. Bechtold, J.B. Reddy, A. Choudhary i inni. Can endoscopic ultrasound predict early rectal cancers that can be resected endoscopically? A meta-analysis and systematic review. „Dig Dis Sci”. 55 (5), s. 1221–1229, maj 2010. DOI: 10.1007/s10620-009-0862-9. PMID: 19517233.
- ↑ S.R. Puli, M.L. Bechtold, J.B. Reddy, A. Choudhary i inni. How good is endoscopic ultrasound in differentiating various T stages of rectal cancer? Meta-analysis and systematic review. „Ann Surg Oncol”. 16 (2), s. 254–265, luty 2009. DOI: 10.1245/s10434-008-0231-5. PMID: 19018597.
- ↑ M.J. Lahaye, S.M. Engelen, P.J. Nelemans, G.L. Beets i inni. Imaging for predicting the risk factors--the circumferential resection margin and nodal disease-of local recurrence in rectal cancer: a meta-analysis. „Semin Ultrasound CT MR”. 26 (4), s. 259–268, sierpień 2005. PMID: 16152740.
- ↑ M.E. Riccioni, R. Urgesi, R. Cianci, A. Bizzotto i inni. Colon capsule endoscopy: Advantages, limitations and expectations. Which novelties?. „World J Gastrointest Endosc”. 4 (4), s. 99–107, kwiecień 2012. DOI: 10.4253/wjge.v4.i4.99. PMID: 22523610.
- ↑ a b C. Spada, F. Barbaro, G. Andrisani, L. Minelli Grazioli i inni. Colon capsule endoscopy: What we know and what we would like to know. „World J Gastroenterol”. 20 (45), s. 16948–16955, grudzień 2014. DOI: 10.3748/wjg.v20.i45.16948. PMID: 25493007.
- ↑ a b M.F. Hale, R. Sidhu, M.E. McAlindon. Capsule endoscopy: current practice and future directions. „World J Gastroenterol”. 20 (24), s. 7752–7759, czerwiec 2014. DOI: 10.3748/wjg.v20.i24.7752. PMID: 24976712.
- ↑ R. Eliakim, K. Yassin, Y. Niv, Y. Metzger i inni. Prospective multicenter performance evaluation of the second-generation colon capsule compared with colonoscopy. „Endoscopy”. 41 (12), s. 1026–1031, grudzień 2009. DOI: 10.1055/s-0029-1215360. PMID: 19967618.
- ↑ C. Spada, C. Hassan, M. Munoz-Navas, H. Neuhaus i inni. Second-generation colon capsule endoscopy compared with colonoscopy. „Gastrointest Endosc”. 74 (3), s. 581–589.e1, wrzesień 2011. DOI: 10.1016/j.gie.2011.03.1125. PMID: 21601200.
- ↑ C. Spada, C. Hassan, J.P. Galmiche, H. Neuhaus i inni. Colon capsule endoscopy: European Society of Gastrointestinal Endoscopy (ESGE) Guideline. „Endoscopy”. 44 (5), s. 527–536, maj 2012. DOI: 10.1055/s-0031-1291717. PMID: 22389230.
- ↑ a b c L. Sun, H. Wu, Y.S. Guan. Colonography by CT, MRI and PET/CT combined with conventional colonoscopy in colorectal cancer screening and staging. „World J Gastroenterol”. 14 (6), s. 853–863, luty 2008. PMID: 18240342.
- ↑ P.J. Pickhardt, C. Hassan, S. Halligan, R. Marmo. Colorectal cancer: CT colonography and colonoscopy for detection-systematic review and meta-analysis. „Radiology”. 259 (2), s. 393–405, maj 2011. DOI: 10.1148/radiol.11101887. PMID: 21415247.
- ↑ a b c d e f g M. Kekelidze, L. D’Errico, M. Pansini, A. Tyndall i inni. Colorectal cancer: current imaging methods and future perspectives for the diagnosis, staging and therapeutic response evaluation. „World J Gastroenterol”. 19 (46), s. 8502–8514, grudzień 2013. DOI: 10.3748/wjg.v19.i46.8502. PMID: 24379567.
- ↑ S. Bipat, M.C. Niekel, E.F. Comans, C.Y. Nio i inni. Imaging modalities for the staging of patients with colorectal cancer. „Neth J Med”. 70 (1), s. 26–34, styczeń 2012. PMID: 22271811.
- ↑ F.M. Zijta, S. Bipat, J. Stoker. Magnetic resonance (MR) colonography in the detection of colorectal lesions: a systematic review of prospective studies. „Eur Radiol”. 20 (5), s. 1031–1046, maj 2010. DOI: 10.1007/s00330-009-1663-4. PMID: 19936754.
- ↑ M. Mulla, R. Deb, R. Singh. MRI in T staging of rectal cancer: How effective is it?. „Indian J Radiol Imaging”. 20 (2), s. 118–121, maj 2010. DOI: 10.4103/0971-3026.63055. PMID: 20607023.
- ↑ U. Tapan, M. Ozbayrak, S. Tatlı. MRI in local staging of rectal cancer: an update. „Diagn Interv Radiol”. 20 (5). s. 390–398. DOI: 10.5152/dir.2014.13265. PMID: 25010367.
- ↑ C. Klessen, P. Rogalla, M. Taupitz. Local staging of rectal cancer: the current role of MRI. „Eur Radiol”. 17 (2), s. 379–389, luty 2007. DOI: 10.1007/s00330-006-0388-x. PMID: 17008990.
- ↑ a b M.C. Niekel, S. Bipat, J. Stoker. Diagnostic imaging of colorectal liver metastases with CT, MR imaging, FDG PET, and/or FDG PET/CT: a meta-analysis of prospective studies including patients who have not previously undergone treatment. „Radiology”. 257 (3), s. 674–684, grudzień 2010. DOI: 10.1148/radiol.10100729. PMID: 20829538.
- ↑ D. Delbeke, W.H. Martin. FDG PET and PET/CT for colorectal cancer. „Methods Mol Biol”. 727, s. 77–103, 2011. DOI: 10.1007/978-1-61779-062-1_6. PMID: 21331930.
- ↑ F.U. Chowdhury, N. Shah, A.F. Scarsbrook, K.M. Bradley. [18F]FDG PET/CT imaging of colorectal cancer: a pictorial review. „Postgrad Med J”. 86 (1013), s. 174–182, marzec 2010. DOI: 10.1136/pgmj.2009.079087. PMID: 20237012.
- ↑ L.F. de Geus-Oei, T.J. Ruers, C.J. Punt, J.W. Leer i inni. FDG-PET in colorectal cancer. „Cancer Imaging”. 6, s. S71-81, 2006. DOI: 10.1102/1470-7330.2006.9014. PMID: 17114081.
- ↑ Bogdan Pruszyński, Bogusława Benendo-Kapuścińska: Radiologia: diagnostyka obrazowa: Rtg, TK, USG, MR i radioizotopy. PZWL, 2008, s. 285. ISBN 978-83-200-3666-4.
- ↑ Vincenza Snow: Screening for Diseases: Prevention in Primary Care. ACP Press, 2004, s. 107. ISBN 978-1-930513-56-3.
- ↑ Bogdan Pruszyński, Bogusława Benendo-Kapuścińska: Radiologia: diagnostyka obrazowa: Rtg, TK, USG, MR i radioizotopy. PZWL, 2008, s. 296–297. ISBN 978-83-200-3666-4.
- ↑ A.I. Filiz, I. Sucullu, Y. Kurt, D.O. Karakas i inni. Persistent high postoperative carcinoembryonic antigen in colorectal cancer patients--is it important?. „Clinics (Sao Paulo)”. 64 (4), s. 287–294, 2009. PMID: 19488584.
- ↑ T. Allende, J.L. García Muñiz, F. Vizoso, J.M. Del Casar i inni. Preoperative serum levels of the carcinoembryonic antigen (CEA) and prognosis in colorectal cancer. „Rev Esp Med Nucl”. 20 (5), s. 358–364, sierpień 2001. PMID: 11470069.
- ↑ S. Wiratkapun, M. Kraemer, F. Seow-Choen, Y.H. Ho i inni. High preoperative serum carcinoembryonic antigen predicts metastatic recurrence in potentially curative colonic cancer: results of a five-year study. „Dis Colon Rectum”. 44 (2), s. 231–235, luty 2001. PMID: 11227940.
- ↑ a b M.J. Duffy. Carcinoembryonic antigen as a marker for colorectal cancer: is it clinically useful?. „Clin Chem”. 47 (4), s. 624–630, kwiecień 2001. PMID: 11274010.
- ↑ L.A. Carriquiry, A. Piñeyro. Should carcinoembryonic antigen be used in the management of patients with colorectal cancer?. „Dis Colon Rectum”. 42 (7), s. 921–929, lipiec 1999. PMID: 10411440.
- ↑ S.Y. Zhang, M. Lin, H.B. Zhang. Diagnostic value of carcinoembryonic antigen and carcinoma antigen 19-9 for colorectal carcinoma. „Int J Clin Exp Pathol”. 8 (8), s. 9404–9409, 2015. PMID: 26464695.
- ↑ R. Partyka, A. Sandelewski, I. Łobejko, J. Kocot i inni. Usefulness of evaluation of soluble fragment cytokeratin 18, carcinoembryonic antigen and gastrointestinal carcinoma-associated antigen in diagnostic of patients with colorectal cancer. „Pol Merkur Lekarski”. 29 (170), s. 128–130, sierpień 2010. PMID: 20842828.
- ↑ Z. Vukobrat-Bijedic, A. Husic-Selimovic, A. Sofic, N. Bijedic i inni. Cancer Antigens (CEA and CA 19-9) as Markers of Advanced Stage of Colorectal Carcinoma. „Med Arch”. 67 (6), s. 397–401, grudzień 2013. DOI: 10.5455/medarh.2013.67.397-401. PMID: 25568506.
- ↑ S.G. Yeo, D.Y. Kim, T.H. Kim, S.Y. Kim i inni. Carbohydrate antigen 19-9 levels associated with pathological responses to preoperative chemoradiotherapy in rectal cancer. „Asian Pac J Cancer Prev”. 15 (13), s. 5383–5387, 2014. PMID: 25041006.
- ↑ J. Stiksma, D.C. Grootendorst, P.W. van der Linden. CA 19-9 as a marker in addition to CEA to monitor colorectal cancer. „Clin Colorectal Cancer”. 13 (4), s. 239–244, grudzień 2014. DOI: 10.1016/j.clcc.2014.09.004. PMID: 25442815.
- ↑ Y. Narita, H. Taniguchi, A. Komori, S. Nitta i inni. CA19-9 level as a prognostic and predictive factor of bevacizumab efficacy in metastatic colorectal cancer patients undergoing oxaliplatin-based chemotherapy. „Cancer Chemother Pharmacol”. 73 (2), s. 409–416, luty 2014. DOI: 10.1007/s00280-013-2367-7. PMID: 24322376.
- ↑ J.B. Lopez, G.P. Royan, M.N. Lakhwani, M. Mahadaven i inni. CA 72-4 compared with CEA and CA 19-9 as a marker of some gastrointestinal malignancies. „Int J Biol Markers”. 14 (3). s. 172–177. PMID: 10569140.
- ↑ a b G. Lindmark, U. Kressner, R. Bergström, B. Glimelius. Limited clinical significance of the serum tumour marker Ca 72-4 in colorectal cancer. „Anticancer Res”. 16 (2). s. 895–898. PMID: 8687147.
- ↑ J. Louhimo, M. Carpelan-Holmström, H. Alfthan, U.H. Stenman i inni. Serum HCG beta, CA 72-4 and CEA are independent prognostic factors in colorectal cancer. „Int J Cancer”. 101 (6), s. 545–548, październik 2002. DOI: 10.1002/ijc.90009. PMID: 12237895.
- ↑ D. Ayude, F.J. Rodríguez-Berrocal, J. Ayude, S. Blanco-Prieto i inni. Preoperative serum CA 72.4 as prognostic factor of recurrence and death, especially at TNM stage II, for colorectal cancer. „BMC Cancer”. 13, s. 543, 2013. DOI: 10.1186/1471-2407-13-543. PMID: 24215576.
- ↑ A. Dembińska-Kieć, J.W. Naskalski: Diagnostyka laboratoryjna z elementami biochemii klinicznej. Edra Urban & Partner, 2002, s. 877–878. ISBN 978-83-87944-33-9.
- ↑ a b M. Mishaeli, B. Klein, E. Sadikov, I. Bayer i inni. Initial TPS serum level as an indicator of relapse and survival in colorectal cancer. „Anticancer Res”. 18 (3B). s. 2101–2105. PMID: 9677475.
- ↑ Willett 2001 ↓, s. 53.
- ↑ a b c Krzakowski i in. 2015 ↓, s. 615.
- ↑ Benson i in. 2015 ↓, s. 44–46.
- ↑ Benson i in. 2015 ↓, s. 43.
- ↑ Labianca i in. 2013 ↓, s. 2.
- ↑ Willett 2001 ↓, s. 2.
- ↑ H.Y. Kim, S.J. Lee, G. Lee, L. Song i inni. Should preoperative chest CT be recommended to all colon cancer patients?. „Ann Surg”. 259 (2), s. 323–328, luty 2014. DOI: 10.1097/SLA.0b013e3182865080. PMID: 23426347.
- ↑ a b c Labianca i in. 2013 ↓, s. 3.
- ↑ Labianca i in. 2013 ↓, s. 3–4.
- ↑ Benson i in. 2015 ↓, s. 7.
- ↑ a b c d I. Zlobec, A. Lugli. Prognostic and predictive factors in colorectal cancer. „Postgrad Med J”. 84 (994), s. 403–411, sierpień 2008. DOI: 10.1136/jcp.2007.054858. PMID: 18832400.
- ↑ a b c d e f g O. Marzouk, J. Schofield. Review of histopathological and molecular prognostic features in colorectal cance. „Cancers (Basel)”. 3 (2), s. 2767–2810, 2011. DOI: 10.3390/cancers3022767. PMID: 24212832.
- ↑ a b c d H. Ueno, H. Mochizuki, Y. Hashiguchi, H. Shimazaki i inni. Risk factors for an adverse outcome in early invasive colorectal carcinoma. „Gastroenterology”. 127 (2), s. 385–394, sierpień 2004. PMID: 15300569.
- ↑ C.C. Compton. Colorectal carcinoma: diagnostic, prognostic, and molecular features. „Mod Pathol”. 16 (4), s. 376–388, kwiecień 2003. DOI: 10.1097/01.MP.0000062859.46942.93. PMID: 12692203.
- ↑ a b c d C. Compton, C.M. Fenoglio-Preiser, N. Pettigrew, L.P. Fielding. American Joint Committee on Cancer Prognostic Factors Consensus Conference: Colorectal Working Group. „Cancer”. 88 (7), s. 1739–1757, kwiecień 2000. PMID: 10738234.
- ↑ C.C. Compton, L.P. Fielding, L.J. Burgart, B. Conley i inni. Prognostic factors in colorectal cancer. College of American Pathologists Consensus Statement 1999. „Arch Pathol Lab Med”. 124 (7), s. 979–994, lipiec 2000. DOI: <0979:PFICC>2.0.CO;2 10.1043/0003-9985(2000)124<0979:PFICC>2.0.CO;2. PMID: 10888773.
- ↑ H. Ueno, A.B. Price, K.H. Wilkinson, J.R. Jass i inni. A new prognostic staging system for rectal cancer. „Ann Surg”. 240 (5), s. 832–839, listopad 2004. PMID: 15492565.
- ↑ Hamilton i Aaltonen 2000 ↓, s. 117.
- ↑ V.H. Koelzer, A. Lugli. The tumor border configuration of colorectal cancer as a histomorphological prognostic indicator. „Front Oncol”. 4, s. 29, 2014. DOI: 10.3389/fonc.2014.00029. PMID: 24600585.
- ↑ R.C. Newland, O.F. Dent, M.N. Lyttle, P.H. Chapuis i inni. Pathologic determinants of survival associated with colorectal cancer with lymph node metastases. A multivariate analysis of 579 patients. „Cancer”. 73 (8), s. 2076–2082, kwiecień 1994. PMID: 8156513.
- ↑ L. Roncucci, R. Fante, L. Losi, C. Di Gregorio i inni. Survival for colon and rectal cancer in a population-based cancer registry. „Eur J Cancer”. 32A (2), s. 295–302, luty 1996. PMID: 8664045.
- ↑ J. Betge, M.J. Pollheimer, R.A. Lindtner, P. Kornprat i inni. Intramural and extramural vascular invasion in colorectal cancer: prognostic significance and quality of pathology reporting. „Cancer”. 118 (3), s. 628–638, luty 2012. DOI: 10.1002/cncr.26310. PMID: 21751188.
- ↑ C. Liebig, G. Ayala, J. Wilks, G. Verstovsek i inni. Perineural invasion is an independent predictor of outcome in colorectal cancer. „J Clin Oncol”. 27 (31), s. 5131–5137, listopad 2009. DOI: 10.1200/JCO.2009.22.4949. PMID: 19738119.
- ↑ a b Benson i in. 2015 ↓, s. 39.
- ↑ a b mp.pl: Rak okrężnicy i odbytnicy. Klasyfikacja TNM. [dostęp 2015-11-12]. [zarchiwizowane z tego adresu (2015-11-12)].
- ↑ a b Kordek 2007 ↓, s. 181–182.
- ↑ Kordek 2007 ↓, s. 182.
- ↑ Janet Husband, Rodney H. Reznek: Imaging in Oncology, Second Edition. CRC Press, 2004, s. 220. ISBN 978-1-4987-1651-2.
- ↑ a b c d e Benson i in. 2015 ↓, s. 48.
- ↑ Enzinger i in. 2015 ↓, s. 17.
- ↑ DeVita, Lawrence i Rosenberg 2008 ↓, s. 1247–1249.
- ↑ DeVita, Lawrence i Rosenberg 2008 ↓, s. 1289–1290.
- ↑ a b c d DeVita, Lawrence i Rosenberg 2008 ↓, s. 1290.
- ↑ a b Enzinger i in. 2015 ↓, s. 8–9.
- ↑ a b c Krzakowski i in. 2015 ↓, s. 631–632.
- ↑ a b Glimelius i in. 2013 ↓, s. 4.
- ↑ Benson i in. 2015 ↓, s. 8.
- ↑ a b c d e f Van Cutsem i in. 2014 ↓, s. 4.
- ↑ a b Benson i in. 2015 ↓, s. 25–28.
- ↑ DeVita, Lawrence i Rosenberg 2008 ↓, s. 1246–1247.
- ↑ a b c Labianca i in. 2013 ↓, s. 5.
- ↑ a b c DeVita, Lawrence i Rosenberg 2008 ↓, s. 1247.
- ↑ T. Sakamoto, G. Mori, M. Yamada, Y. Kinjo i inni. Endoscopic submucosal dissection for colorectal neoplasms: a review. „World J Gastroenterol”. 20 (43), s. 16153–16158, listopad 2014. DOI: 10.3748/wjg.v20.i43.16153. PMID: 25473168.
- ↑ S.L. Bosch, S. Teerenstra, J.H. de Wilt, C. Cunningham i inni. Predicting lymph node metastasis in pT1 colorectal cancer: a systematic review of risk factors providing rationale for therapy decisions. „Endoscopy”. 45 (10), s. 827–834, październik 2013. DOI: 10.1055/s-0033-1344238. PMID: 23884793.
- ↑ a b c Benson i in. 2015 ↓, s. 49.
- ↑ Welch, Ottinger i Welch 2012 ↓, s. 48.
- ↑ a b c Ronnie Ann Rosenthal, Michael E. Zenilman, Mark R. Katlic: Principles and Practice of Geriatric Surgery. Springer Science & Business Media, 2011, s. 899. ISBN 978-1-4419-6999-6.
- ↑ a b c d John L. Cameron, Andrew M. Cameron: Current Surgical Therapy. Elsevier Health Sciences, 2013, s. 214. ISBN 978-0-323-22511-3.
- ↑ Joe Tjandra, Gordon J.A. Clunie, Andrew H. Kaye, Julian A. Smith: Textbook of Surgery. John Wiley & Sons, 2008, s. 197. ISBN 978-0-470-75779-6.
- ↑ a b DeVita, Lawrence i Rosenberg 2008 ↓, s. 1248.
- ↑ Welch, Ottinger i Welch 2012 ↓, s. 54–55.
- ↑ a b Welch, Ottinger i Welch 2012 ↓, s. 55.
- ↑ a b Ronnie Ann Rosenthal, Michael E. Zenilman, Mark R. Katlic: Principles and Practice of Geriatric Surgery. Springer Science & Business Media, 2011, s. 900. ISBN 978-1-4419-6999-6.
- ↑ Welch, Ottinger i Welch 2012 ↓, s. 56.
- ↑ DeVita, Lawrence i Rosenberg 2008 ↓, s. 1249.
- ↑ Welch, Ottinger i Welch 2012 ↓, s. 58.
- ↑ a b c d e f Roman Herman, Jarosław Reguła, Jakub Pałucki, Wojciech Polkowski, Piotr Potemski: Rak odbytnicy. 2014.
- ↑ a b c d Enzinger i in. 2015 ↓, s. 51.
- ↑ a b c Enzinger i in. 2015 ↓, s. 49.
- ↑ DeVita, Lawrence i Rosenberg 2008 ↓, s. 1288–1289.
- ↑ C.G. Willett, C.C. Compton, P.C. Shellito, J.T. Efird. Selection factors for local excision or abdominoperineal resection of early stage rectal cancer. „Cancer”. 73 (11), s. 2716–2720, czerwiec 1994. PMID: 8194011.
- ↑ a b c d T.A. Heafner, S.C. Glasgow. A critical review of the role of local excision in the treatment of early (T1 and T2) rectal tumors. „J Gastrointest Oncol”. 5 (5), s. 345–352, październik 2014. DOI: 10.3978/j.issn.2078-6891.2014.066. PMID: 25276407.
- ↑ D. Hahnloser, B.G. Wolff, D.W. Larson, J. Ping i inni. Immediate radical resection after local excision of rectal cancer: an oncologic compromise?. „Dis Colon Rectum”. 48 (3), s. 429–437, marzec 2005. DOI: 10.1007/s10350-004-0900-9. PMID: 15747069.
- ↑ a b c d DeVita, Lawrence i Rosenberg 2008 ↓, s. 1289.
- ↑ a b c d Enzinger i in. 2015 ↓, s. 50.
- ↑ D.J. Bentrem, S. Okabe, W.D. Wong, J.G. Guillem i inni. T1 adenocarcinoma of the rectum: transanal excision or radical surgery?. „Ann Surg”. 242 (4), s. 472–477; discussion 477-9, październik 2005. PMID: 16192807.
- ↑ R. Nascimbeni, L.J. Burgart, S. Nivatvongs, D.R. Larson. Risk of lymph node metastasis in T1 carcinoma of the colon and rectum. „Dis Colon Rectum”. 45 (2), s. 200–206, luty 2002. PMID: 11852333.
- ↑ Y.N. You, N.N. Baxter, A. Stewart, H. Nelson. Is the increasing rate of local excision for stage I rectal cancer in the United States justified?: a nationwide cohort study from the National Cancer Database. „Ann Surg”. 245 (5), s. 726–733, maj 2007. DOI: 10.1097/01.sla.0000252590.95116.4f. PMID: 17457165.
- ↑ G.M. Nash, M.R. Weiser, J.G. Guillem, L.K. Temple i inni. Long-term survival after transanal excision of T1 rectal cancer. „Dis Colon Rectum”. 52 (4), s. 577–582, kwiecień 2009. DOI: 10.1007/DCR.0b013e3181a0adbd. PMID: 19404055.
- ↑ K.B. Stitzenberg, H.K. Sanoff, D.C. Penn, M.O. Meyers i inni. Practice patterns and long-term survival for early-stage rectal cancer. „J Clin Oncol”. 31 (34), s. 4276–4282, grudzień 2013. DOI: 10.1200/JCO.2013.49.1860. PMID: 24166526.
- ↑ Kelsen 2008 ↓, s. 596.
- ↑ Jeffrey Norton, R.Randall Bollinger, Alfred E. Chang, Stephen F. Lowry: Surgery: Basic Science and Clinical Evidence. Springer, 2012, s. 717. ISBN 978-3-642-57282-1.
- ↑ Willett 2001 ↓, s. 112.
- ↑ E. Tiret, B. Poupardin, D. McNamara, N. Dehni i inni. Ultralow anterior resection with intersphincteric dissection-what is the limit of safe sphincter preservation?. „Colorectal Dis”. 5 (5), s. 454–457, wrzesień 2003. PMID: 12925080.
- ↑ M. den Dulk, H. Putter, L. Collette, C.A. Marijnen i inni. The abdominoperineal resection itself is associated with an adverse outcome: the European experience based on a pooled analysis of five European randomised clinical trials on rectal cancer. „Eur J Cancer”. 45 (7), s. 1175–1183, maj 2009. DOI: 10.1016/j.ejca.2008.11.039. PMID: 19128956.
- ↑ L. Påhlman, M. Bohe, B. Cedermark, M. Dahlberg i inni. The Swedish rectal cancer registry. „Br J Surg”. 94 (10), s. 1285–1292, październik 2007. DOI: 10.1002/bjs.5679. PMID: 17661309.
- ↑ R.O. Lindsetmo, Y.G. Joh, C.P. Delaney. Surgical treatment for rectal cancer: an international perspective on what the medical gastroenterologist needs to know. „World J Gastroenterol”. 14 (21), s. 3281–3289, czerwiec 2008. PMID: 18528924.
- ↑ Krzysztof Bujko, Marek P. Nowacki. Napromienianie uzupełniające chorych na raka odbytnicy. „Nowa Medycyna”, 1999.
- ↑ Bartłomiej Szynglarewicz, Rafał Matkowski, Daniel Sydor, Józef Forgacz i inni. Skuteczność resekcji przedniej z całkowitym wycięciem mezorektum w radykalnym leczeniu raka odbytnicy u mężczyzn. „Współczesna Onkologia”, 2007.
- ↑ A. Martling, T. Holm, L.E. Rutqvist, H. Johansson i inni. Impact of a surgical training programme on rectal cancer outcomes in Stockholm. „Br J Surg”. 92 (2), s. 225–229, luty 2005. DOI: 10.1002/bjs.4834. PMID: 15609382.
- ↑ N. Scott, P. Jackson, T. al-Jaberi, M.F. Dixon i inni. Total mesorectal excision and local recurrence: a study of tumour spread in the mesorectum distal to rectal cancer. „Br J Surg”. 82 (8), s. 1031–1033, sierpień 1995. PMID: 7648142.
- ↑ P.T. Phang. Total mesorectal excision: technical aspects. „Can J Surg”. 47 (2), s. 130–137, kwiecień 2004. PMID: 15132469.
- ↑ W.E. Enker, H.T. Thaler, M.L. Cranor, T. Polyak. Total mesorectal excision in the operative treatment of carcinoma of the rectum. „J Am Coll Surg”. 181 (4), s. 335–346, październik 1995. PMID: 7551328.
- ↑ A.M. Lacy, J.C. García-Valdecasas, S. Delgado, A. Castells i inni. Laparoscopy-assisted colectomy versus open colectomy for treatment of non-metastatic colon cancer: a randomised trial. „Lancet”. 359 (9325), s. 2224–2229, czerwiec 2002. DOI: 10.1016/S0140-6736(02)09290-5. PMID: 12103285.
- ↑ M. Buunen, R. Veldkamp, W.C. Hop, E. Kuhry i inni. Survival after laparoscopic surgery versus open surgery for colon cancer: long-term outcome of a randomised clinical trial. „Lancet Oncol”. 10 (1), s. 44–52, styczeń 2009. DOI: 10.1016/S1470-2045(08)70310-3. PMID: 19071061.
- ↑ B.L. Green, H.C. Marshall, F. Collinson, P. Quirke i inni. Long-term follow-up of the Medical Research Council CLASICC trial of conventional versus laparoscopically assisted resection in colorectal cancer. „Br J Surg”. 100 (1), s. 75–82, styczeń 2013. DOI: 10.1002/bjs.8945. PMID: 23132548.
- ↑ a b H. Ohtani, Y. Tamamori, Y. Arimoto, Y. Nishiguchi i inni. A meta-analysis of the short- and long-term results of randomized controlled trials that compared laparoscopy-assisted and open colectomy for colon cancer. „J Cancer”. 3, s. 49–57, 2012. DOI: 10.7150/jca.3621. PMID: 22315650.
- ↑ B. Di, Y. Li, K. Wei, X. Xiao i inni. Laparoscopic versus open surgery for colon cancer: a meta-analysis of 5-year follow-up outcomes. „Surg Oncol”. 22 (3), s. e39-43, wrzesień 2013. DOI: 10.1016/j.suronc.2013.03.002. PMID: 23643698.
- ↑ E. Kuhry, W. Schwenk, R. Gaupset, U. Romild i inni. Long-term outcome of laparoscopic surgery for colorectal cancer: a cochrane systematic review of randomised controlled trials. „Cancer Treat Rev”. 34 (6), s. 498–504, październik 2008. DOI: 10.1016/j.ctrv.2008.03.011. PMID: 18468803.
- ↑ F. Rondelli, S. Trastulli, N. Avenia, G. Schillaci i inni. Is laparoscopic right colectomy more effective than open resection? A meta-analysis of randomized and nonrandomized studies. „Colorectal Dis”. 14 (8), s. e447-69, sierpień 2012. DOI: 10.1111/j.1463-1318.2012.03054.x. PMID: 22540533.
- ↑ Benson i in. 2015 ↓, s. 50.
- ↑ D.G. Jayne, H.C. Thorpe, J. Copeland, P. Quirke i inni. Five-year follow-up of the Medical Research Council CLASICC trial of laparoscopically assisted versus open surgery for colorectal cancer. „Br J Surg”. 97 (11), s. 1638–1645, listopad 2010. DOI: 10.1002/bjs.7160. PMID: 20629110.
- ↑ D.G. Jayne, P.J. Guillou, H. Thorpe, P. Quirke i inni. Randomized trial of laparoscopic-assisted resection of colorectal carcinoma: 3-year results of the UK MRC CLASICC Trial Group. „J Clin Oncol”. 25 (21), s. 3061–3068, lipiec 2007. DOI: 10.1200/JCO.2006.09.7758. PMID: 17634484.
- ↑ D.P. Nussbaum, P.J. Speicher, A.M. Ganapathi, B.R. Englum i inni. Laparoscopic versus open low anterior resection for rectal cancer: results from the national cancer data base. „J Gastrointest Surg”. 19 (1), s. 124–131; discussion 131-2, styczeń 2015. DOI: 10.1007/s11605-014-2614-1. PMID: 25091847.
- ↑ Enzinger i in. 2015 ↓, s. 51–52.
- ↑ Benson i in. 2015 ↓, s. 50–51.
- ↑ Benson i in. 2015 ↓, s. 51.
- ↑ a b c Benson i in. 2015 ↓, s. 52.
- ↑ a b S. Gill, C.L. Loprinzi, D.J. Sargent, S.D. Thomé i inni. Pooled analysis of fluorouracil-based adjuvant therapy for stage II and III colon cancer: who benefits and by how much?. „J Clin Oncol”. 22 (10), s. 1797–1806, maj 2004. DOI: 10.1200/JCO.2004.09.059. PMID: 15067028.
- ↑ R. Gray, J. Barnwell, C. McConkey, R.K. Hills i inni. Adjuvant chemotherapy versus observation in patients with colorectal cancer: a randomised study. „Lancet”. 370 (9604), s. 2020–2029, grudzień 2007. DOI: 10.1016/S0140-6736(07)61866-2. PMID: 18083404.
- ↑ X. Wu, J. Zhang, X. He, C. Wang i inni. Postoperative adjuvant chemotherapy for stage II colorectal cancer: a systematic review of 12 randomized controlled trials. „J Gastrointest Surg”. 16 (3), s. 646–655, marzec 2012. DOI: 10.1007/s11605-011-1682-8. PMID: 22194062.
- ↑ E.S. O’Connor, D.Y. Greenblatt, N.K. LoConte, R.E. Gangnon i inni. Adjuvant chemotherapy for stage II colon cancer with poor prognostic features. „J Clin Oncol”. 29 (25), s. 3381–3388, wrzesień 2011. DOI: 10.1200/JCO.2010.34.3426. PMID: 21788561.
- ↑ J.J. Biagi, M.J. Raphael, W.J. Mackillop, W. Kong i inni. Association between time to initiation of adjuvant chemotherapy and survival in colorectal cancer: a systematic review and meta-analysis. „JAMA”. 305 (22), s. 2335–2342, czerwiec 2011. DOI: 10.1001/jama.2011.749. PMID: 21642686.
- ↑ Benson i in. 2015 ↓, s. 54.
- ↑ a b Benson i in. 2015 ↓, s. 56.
- ↑ T. André, C. Boni, M. Navarro, J. Tabernero i inni. Improved overall survival with oxaliplatin, fluorouracil, and leucovorin as adjuvant treatment in stage II or III colon cancer in the MOSAIC trial. „J Clin Oncol”. 27 (19), s. 3109–3116, lipiec 2009. DOI: 10.1200/JCO.2008.20.6771. PMID: 19451431.
- ↑ H.K. Sanoff, W.R. Carpenter, C.F. Martin, D.J. Sargent i inni. Comparative effectiveness of oxaliplatin vs non-oxaliplatin-containing adjuvant chemotherapy for stage III colon cancer. „J Natl Cancer Inst”. 104 (3), s. 211–227, luty 2012. DOI: 10.1093/jnci/djr524. PMID: 22266473.
- ↑ H.K. Sanoff, W.R. Carpenter, J. Freburger, L. Li i inni. Comparison of adverse events during 5-fluorouracil versus 5-fluorouracil/oxaliplatin adjuvant chemotherapy for stage III colon cancer: a population-based analysis. „Cancer”. 118 (17), s. 4309–4320, wrzesień 2012. DOI: 10.1002/cncr.27422. PMID: 22294436.
- ↑ J.P. Kuebler, H.S. Wieand, M.J. O’Connell, R.E. Smith i inni. Oxaliplatin combined with weekly bolus fluorouracil and leucovorin as surgical adjuvant chemotherapy for stage II and III colon cancer: results from NSABP C-07. „J Clin Oncol”. 25 (16), s. 2198–2204, czerwiec 2007. DOI: 10.1200/JCO.2006.08.2974. PMID: 17470851.
- ↑ G. Yothers, M.J. O’Connell, C.J. Allegra, J.P. Kuebler i inni. Oxaliplatin as adjuvant therapy for colon cancer: updated results of NSABP C-07 trial, including survival and subset analyses. „J Clin Oncol”. 29 (28), s. 3768–3774, październik 2011. DOI: 10.1200/JCO.2011.36.4539. PMID: 21859995.
- ↑ a b c Benson i in. 2015 ↓, s. 57.
- ↑ S. Sharif, M.J. O’Connell, G. Yothers, S. Lopa i inni. FOLFOX and FLOX regimens for the adjuvant treatment of resected stage II and III colon cancer. „Cancer Invest”. 26 (9), s. 956–963, listopad 2008. DOI: 10.1080/07357900802132550. PMID: 18798075.
- ↑ C. Twelves, A. Wong, M.P. Nowacki, M. Abt i inni. Capecitabine as adjuvant treatment for stage III colon cancer. „N Engl J Med”. 352 (26), s. 2696–2704, czerwiec 2005. DOI: 10.1056/NEJMoa043116. PMID: 15987918.
- ↑ C. Twelves, W. Scheithauer, J. McKendrick, J.F. Seitz i inni. Capecitabine versus 5-fluorouracil/folinic acid as adjuvant therapy for stage III colon cancer: final results from the X-ACT trial with analysis by age and preliminary evidence of a pharmacodynamic marker of efficacy. „Ann Oncol”. 23 (5), s. 1190–1197, maj 2012. DOI: 10.1093/annonc/mdr366. PMID: 21896539.
- ↑ D.G. Haller, J. Tabernero, J. Maroun, F. de Braud i inni. Capecitabine plus oxaliplatin compared with fluorouracil and folinic acid as adjuvant therapy for stage III colon cancer. „J Clin Oncol”. 29 (11), s. 1465–1471, kwiecień 2011. DOI: 10.1200/JCO.2010.33.6297. PMID: 21383294.
- ↑ a b Benson i in. 2015 ↓, s. 58.
- ↑ a b Enzinger i in. 2015 ↓, s. 52.
- ↑ Enzinger i in. 2015 ↓, s. 7–10.
- ↑ a b c d e f Enzinger i in. 2015 ↓, s. 61.
- ↑ Enzinger i in. 2015 ↓, s. 8.
- ↑ a b L.L. Gunderson, D.J. Sargent, J.E. Tepper, N. Wolmark i inni. Impact of T and N stage and treatment on survival and relapse in adjuvant rectal cancer: a pooled analysis. „J Clin Oncol”. 22 (10), s. 1785–1796, maj 2004. DOI: 10.1200/JCO.2004.08.173. PMID: 15067027.
- ↑ a b Enzinger i in. 2015 ↓, s. 9.
- ↑ Enzinger i in. 2015 ↓, s. 10.
- ↑ Krzakowski i in. 2015 ↓, s. 634.
- ↑ Krzakowski i in. 2015 ↓, s. 632.
- ↑ a b c Enzinger i in. 2015 ↓, s. 53.
- ↑ a b R. Sauer, H. Becker, W. Hohenberger, C. Rödel i inni. Preoperative versus postoperative chemoradiotherapy for rectal cancer. „N Engl J Med”. 351 (17), s. 1731–1740, październik 2004. DOI: 10.1056/NEJMoa040694. PMID: 15496622.
- ↑ R. Wagman, B.D. Minsky, A.M. Cohen, J.G. Guillem i inni. Sphincter preservation in rectal cancer with preoperative radiation therapy and coloanal anastomosis: long term follow-up. „Int J Radiat Oncol Biol Phys”. 42 (1), s. 51–57, sierpień 1998. PMID: 9747819.
- ↑ R. Sauer, T. Liersch, S. Merkel, R. Fietkau i inni. Preoperative versus postoperative chemoradiotherapy for locally advanced rectal cancer: results of the German CAO/ARO/AIO-94 randomized phase III trial after a median follow-up of 11 years. „J Clin Oncol”. 30 (16), s. 1926–1933, czerwiec 2012. DOI: 10.1200/JCO.2011.40.1836. PMID: 22529255.
- ↑ L.C. Peng, J. Milsom, K. Garrett, G. Nandakumar i inni. Surveillance, epidemiology, and end results-based analysis of the impact of preoperative or postoperative radiotherapy on survival outcomes for T3N0 rectal cancer. „Cancer Epidemiol”. 38 (1), s. 73–78, luty 2014. DOI: 10.1016/j.canep.2013.12.008. PMID: 24491755.
- ↑ a b Enzinger i in. 2015 ↓, s. 57.
- ↑ J.P. Gérard, T. Conroy, F. Bonnetain, O. Bouché i inni. Preoperative radiotherapy with or without concurrent fluorouracil and leucovorin in T3-4 rectal cancers: results of FFCD 9203. „J Clin Oncol”. 24 (28), s. 4620–4625, październik 2006. DOI: 10.1200/JCO.2006.06.7629. PMID: 17008704.
- ↑ J.F. Bosset, G. Calais, L. Mineur, P. Maingon i inni. Enhanced tumorocidal effect of chemotherapy with preoperative radiotherapy for rectal cancer: preliminary results-EORTC 22921. „J Clin Oncol”. 23 (24), s. 5620–5627, sierpień 2005. DOI: 10.1200/JCO.2005.02.113. PMID: 16009958.
- ↑ a b J.F. Bosset, L. Collette, G. Calais, L. Mineur i inni. Chemotherapy with preoperative radiotherapy in rectal cancer. „N Engl J Med”. 355 (11), s. 1114–1123, wrzesień 2006. DOI: 10.1056/NEJMoa060829. PMID: 16971718.
- ↑ W.P. Ceelen, Y. Van Nieuwenhove, K. Fierens. Preoperative chemoradiation versus radiation alone for stage II and III resectable rectal cancer. „Cochrane Database Syst Rev”, s. CD006041, 2009. DOI: 10.1002/14651858.CD006041.pub2. PMID: 19160264.
- ↑ K. McCarthy, K. Pearson, R. Fulton, J. Hewitt. Pre-operative chemoradiation for non-metastatic locally advanced rectal cancer. „Cochrane Database Syst Rev”. 12, s. CD008368, 2012. DOI: 10.1002/14651858.CD008368.pub2. PMID: 23235660.
- ↑ N.N. Rahbari, H. Elbers, V. Askoxylakis, E. Motschall i inni. Neoadjuvant radiotherapy for rectal cancer: meta-analysis of randomized controlled trials. „Ann Surg Oncol”. 20 (13), s. 4169–4182, grudzień 2013. DOI: 10.1245/s10434-013-3198-9. PMID: 24002536.
- ↑ a b M.J. O’Connell, L.H. Colangelo, R.W. Beart, N.J. Petrelli i inni. Capecitabine and oxaliplatin in the preoperative multimodality treatment of rectal cancer: surgical end points from National Surgical Adjuvant Breast and Bowel Project trial R-04. „J Clin Oncol”. 32 (18), s. 1927–1934, czerwiec 2014. DOI: 10.1200/JCO.2013.53.7753. PMID: 24799484.
- ↑ R.D. Hofheinz, F. Wenz, S. Post, A. Matzdorff i inni. Chemoradiotherapy with capecitabine versus fluorouracil for locally advanced rectal cancer: a randomised, multicentre, non-inferiority, phase 3 trial. „Lancet Oncol”. 13 (6), s. 579–588, czerwiec 2012. DOI: 10.1016/S1470-2045(12)70116-X. PMID: 22503032.
- ↑ C. Aschele, L. Cionini, S. Lonardi, C. Pinto i inni. Primary tumor response to preoperative chemoradiation with or without oxaliplatin in locally advanced rectal cancer: pathologic results of the STAR-01 randomized phase III trial. „J Clin Oncol”. 29 (20), s. 2773–2780, lipiec 2011. DOI: 10.1200/JCO.2010.34.4911. PMID: 21606427.
- ↑ C. Rödel, T. Liersch, H. Becker, R. Fietkau i inni. Preoperative chemoradiotherapy and postoperative chemotherapy with fluorouracil and oxaliplatin versus fluorouracil alone in locally advanced rectal cancer: initial results of the German CAO/ARO/AIO-04 randomised phase 3 trial. „Lancet Oncol”. 13 (7), s. 679–687, lipiec 2012. DOI: 10.1016/S1470-2045(12)70187-0. PMID: 22627104.
- ↑ J.P. Gérard, D. Azria, S. Gourgou-Bourgade, I. Martel-Lafay i inni. Clinical outcome of the ACCORD 12/0405 PRODIGE 2 randomized trial in rectal cancer. „J Clin Oncol”. 30 (36), s. 4558–4565, grudzień 2012. DOI: 10.1200/JCO.2012.42.8771. PMID: 23109696.
- ↑ K. Bujko, M.P. Nowacki, A. Nasierowska-Guttmejer, W. Michalski i inni. Long-term results of a randomized trial comparing preoperative short-course radiotherapy with preoperative conventionally fractionated chemoradiation for rectal cancer. „Br J Surg”. 93 (10), s. 1215–1223, październik 2006. DOI: 10.1002/bjs.5506. PMID: 16983741.
- ↑ S. Ngan, R. Fisher, D. Goldstein, M. Solomon i inni. A randomized trial comparing local recurrence (LR) rates between short-course (SC) and long-course (LC) preoperative radiotherapy (RT) for clinical T3 rectal cancer: An intergroup trial (TROG, AGITG, CSSANZ, RACS). „J Clin Oncol”, 2010.
- ↑ S.Y. Ngan, B. Burmeister, R.J. Fisher, M. Solomon i inni. Randomized trial of short-course radiotherapy versus long-course chemoradiation comparing rates of local recurrence in patients with T3 rectal cancer: Trans-Tasman Radiation Oncology Group trial 01.04. „J Clin Oncol”. 30 (31), s. 3827–3833, listopad 2012. DOI: 10.1200/JCO.2012.42.9597. PMID: 23008301.
- ↑ T. Latkauskas, H. Pauzas, I. Gineikiene, R. Janciauskiene i inni. Initial results of a randomized controlled trial comparing clinical and pathological downstaging of rectal cancer after preoperative short-course radiotherapy or long-term chemoradiotherapy, both with delayed surgery. „Colorectal Dis”. 14 (3), s. 294–298, marzec 2012. DOI: 10.1111/j.1463-1318.2011.02815.x. PMID: 21899712.
- ↑ Enzinger i in. 2015 ↓, s. 56.
- ↑ C. Fernández-Martos, C. Pericay, J. Aparicio, A. Salud i inni. Phase II, randomized study of concomitant chemoradiotherapy followed by surgery and adjuvant capecitabine plus oxaliplatin (CAPOX) compared with induction CAPOX followed by concomitant chemoradiotherapy and surgery in magnetic resonance imaging-defined, locally advanced rectal cancer: Grupo cancer de recto 3 study. „J Clin Oncol”. 28 (5), s. 859–865, luty 2010. DOI: 10.1200/JCO.2009.25.8541. PMID: 20065174.
- ↑ R. Maréchal, B. Vos, M. Polus, T. Delaunoit i inni. Short course chemotherapy followed by concomitant chemoradiotherapy and surgery in locally advanced rectal cancer: a randomized multicentric phase II study. „Ann Oncol”. 23 (6), s. 1525–1530, czerwiec 2012. DOI: 10.1093/annonc/mdr473. PMID: 22039087.
- ↑ a b c Enzinger i in. 2015 ↓, s. 59.
- ↑ Enzinger i in. 2015 ↓, s. 60.
- ↑ J.F. Bosset, G. Calais, L. Mineur, P. Maingon i inni. Fluorouracil-based adjuvant chemotherapy after preoperative chemoradiotherapy in rectal cancer: long-term results of the EORTC 22921 randomised study. „Lancet Oncol”. 15 (2), s. 184–190, luty 2014. DOI: 10.1016/S1470-2045(13)70599-0. PMID: 24440473.
- ↑ S.H. Petersen, H. Harling, L.T. Kirkeby, P. Wille-Jørgensen i inni. Postoperative adjuvant chemotherapy in rectal cancer operated for cure. „Cochrane Database Syst Rev”. 3, s. CD004078, 2012. DOI: 10.1002/14651858.CD004078.pub2. PMID: 22419291.
- ↑ Y.S. Hong, B.H. Nam, K.P. Kim, J.E. Kim i inni. Oxaliplatin, fluorouracil, and leucovorin versus fluorouracil and leucovorin as adjuvant chemotherapy for locally advanced rectal cancer after preoperative chemoradiotherapy (ADORE): an open-label, multicentre, phase 2, randomised controlled trial. „Lancet Oncol”. 15 (11), s. 1245–1253, październik 2014. DOI: 10.1016/S1470-2045(14)70377-8. PMID: 25201358.
- ↑ a b c M.D. Hellinger, C.A. Santiago. Reoperation for recurrent colorectal cancer. „Clin Colon Rectal Surg”. 19 (4), s. 228–236, listopad 2006. DOI: 10.1055/s-2006-956445. PMID: 20011326.
- ↑ a b K.F. Birbeck, C.P. Macklin, N.J. Tiffin, W. Parsons i inni. Rates of circumferential resection margin involvement vary between surgeons and predict outcomes in rectal cancer surgery. „Ann Surg”. 235 (4), s. 449–457, kwiecień 2002. PMID: 11923599.
- ↑ a b c D. Sargent, A. Sobrero, A. Grothey, M.J. O’Connell i inni. Evidence for cure by adjuvant therapy in colon cancer: observations based on individual patient data from 20,898 patients on 18 randomized trials. „J Clin Oncol”. 27 (6), s. 872–877, luty 2009. DOI: 10.1200/JCO.2008.19.5362. PMID: 19124803.
- ↑ S. Galandiuk, H.S. Wieand, C.G. Moertel, S.S. Cha i inni. Patterns of recurrence after curative resection of carcinoma of the colon and rectum. „Surg Gynecol Obstet”. 174 (1), s. 27–32, styczeń 1992. PMID: 1729745.
- ↑ a b D.J. Sargent, H.S. Wieand, D.G. Haller, R. Gray i inni. Disease-free survival versus overall survival as a primary end point for adjuvant colon cancer studies: individual patient data from 20,898 patients on 18 randomized trials. „J Clin Oncol”. 23 (34), s. 8664–8670, grudzień 2005. DOI: 10.1200/JCO.2005.01.6071. PMID: 16260700.
- ↑ DeVita, Lawrence i Rosenberg 2008 ↓, s. 1260.
- ↑ Benson i in. 2015 ↓, s. 14.
- ↑ Van Cutsem i in. 2014 ↓, s. 3–4.
- ↑ Benson i in. 2015 ↓, s. 11.
- ↑ Van Cutsem i in. 2014 ↓, s. 4–5.
- ↑ Van Cutsem i in. 2014 ↓, s. 5.
- ↑ a b Benson i in. 2015 ↓, s. 59–60.
- ↑ Benson i in. 2015 ↓, s. 85.
- ↑ Ö. Akgül, E. Çetinkaya, Ş. Ersöz, M. Tez. Role of surgery in colorectal cancer liver metastases. „World J Gastroenterol”. 20 (20), s. 6113–6122, maj 2014. DOI: 10.3748/wjg.v20.i20.6113. PMID: 24876733.
- ↑ Krzakowski i in. 2015 ↓, s. 618.
- ↑ a b F.I. Macedo, T. Makarawo. Colorectal hepatic metastasis: Evolving therapies. „World J Hepatol”. 6 (7), s. 453–463, lipiec 2014. DOI: 10.4254/wjh.v6.i7.453. PMID: 25067997.
- ↑ S. Vatandoust, T.J. Price, C.S. Karapetis. Colorectal cancer: Metastases to a single organ. „World J Gastroenterol”. 21 (41), s. 11767–11776, listopad 2015. DOI: 10.3748/wjg.v21.i41.11767. PMID: 26557001.
- ↑ G.P. Kanas, A. Taylor, J.N. Primrose, W.J. Langeberg i inni. Survival after liver resection in metastatic colorectal cancer: review and meta-analysis of prognostic factors. „Clin Epidemiol”. 4, s. 283–301, 2012. DOI: 10.2147/CLEP.S34285. PMID: 23152705.
- ↑ M.A. Choti, J.V. Sitzmann, M.F. Tiburi, W. Sumetchotimetha i inni. Trends in long-term survival following liver resection for hepatic colorectal metastases. „Ann Surg”. 235 (6), s. 759–766, czerwiec 2002. PMID: 12035031.
- ↑ T.M. Pawlik, C.R. Scoggins, D. Zorzi, E.K. Abdalla i inni. Effect of surgical margin status on survival and site of recurrence after hepatic resection for colorectal metastases. „Ann Surg”. 241 (5), s. 715–722, discussion 722-4, maj 2005. PMID: 15849507.
- ↑ a b Benson i in. 2015 ↓, s. 59.
- ↑ T.A. Aloia, J.N. Vauthey, E.M. Loyer, D. Ribero i inni. Solitary colorectal liver metastasis: resection determines outcome. „Arch Surg”. 141 (5), s. 460–466; discussion 466-7, maj 2006. DOI: 10.1001/archsurg.141.5.460. PMID: 16702517.
- ↑ W.S. Lee, S.H. Yun, H.K. Chun, W.Y. Lee i inni. Clinical outcomes of hepatic resection and radiofrequency ablation in patients with solitary colorectal liver metastasis. „J Clin Gastroenterol”. 42 (8), s. 945–949, wrzesień 2008. DOI: 10.1097/MCG.0b013e318064e752. PMID: 18438208.
- ↑ H. Hur, Y.T. Ko, B.S. Min, K.S. Kim i inni. Comparative study of resection and radiofrequency ablation in the treatment of solitary colorectal liver metastases. „Am J Surg”. 197 (6), s. 728–736, czerwiec 2009. DOI: 10.1016/j.amjsurg.2008.04.013. PMID: 18789428.
- ↑ H.K. Kim, J.H. Cho, H.Y. Lee, J. Lee i inni. Pulmonary metastasectomy for colorectal cancer: how many nodules, how many times?. „World J Gastroenterol”. 20 (20), s. 6133–6145, maj 2014. DOI: 10.3748/wjg.v20.i20.6133. PMID: 24876735.
- ↑ T. Treasure. Pulmonary Metastasectomy for Colorectal Cancer: Recent Reports Prompt a Review of the Available Evidence. „Curr Colorectal Cancer Rep”. 10 (3), s. 296–302, 2014. DOI: 10.1007/s11888-014-0234-5. PMID: 25191154.
- ↑ C. Pulitanò, M. Bodingbauer, L. Aldrighetti, M.C. de Jong i inni. Liver resection for colorectal metastases in presence of extrahepatic disease: results from an international multi-institutional analysis. „Ann Surg Oncol”. 18 (5), s. 1380–1388, maj 2011. DOI: 10.1245/s10434-010-1459-4. PMID: 21136180.
- ↑ T.C. Chua, A. Saxena, W. Liauw, F. Chu i inni. Hepatectomy and resection of concomitant extrahepatic disease for colorectal liver metastases-a systematic review. „Eur J Cancer”. 48 (12), s. 1757–1765, sierpień 2012. DOI: 10.1016/j.ejca.2011.10.034. PMID: 22153217.
- ↑ D.R. Carpizo, M. D’Angelica. Liver resection for metastatic colorectal cancer in the presence of extrahepatic disease. „Ann Surg Oncol”. 16 (9), s. 2411–2421, wrzesień 2009. DOI: 10.1245/s10434-009-0493-6. PMID: 19554376.
- ↑ D.R. Carpizo, C. Are, W. Jarnagin, R. Dematteo i inni. Liver resection for metastatic colorectal cancer in patients with concurrent extrahepatic disease: results in 127 patients treated at a single center. „Ann Surg Oncol”. 16 (8), s. 2138–2146, sierpień 2009. DOI: 10.1245/s10434-009-0521-6. PMID: 19495884.
- ↑ Benson i in. 2015 ↓, s. 63.
- ↑ Benson i in. 2015 ↓, s. 12.
- ↑ S.R. Alberts, W.L. Horvath, W.C. Sternfeld, R.M. Goldberg i inni. Oxaliplatin, fluorouracil, and leucovorin for patients with unresectable liver-only metastases from colorectal cancer: a North Central Cancer Treatment Group phase II study. „J Clin Oncol”. 23 (36), s. 9243–9249, grudzień 2005. DOI: 10.1200/JCO.2005.07.740. PMID: 16230673.
- ↑ R. Adam, V. Delvart, G. Pascal, A. Valeanu i inni. Rescue surgery for unresectable colorectal liver metastases downstaged by chemotherapy: a model to predict long-term survival. „Ann Surg”. 240 (4), s. 644–657; discussion 657-8, październik 2004. PMID: 15383792.
- ↑ a b A. Falcone, S. Ricci, I. Brunetti, E. Pfanner i inni. Phase III trial of infusional fluorouracil, leucovorin, oxaliplatin, and irinotecan (FOLFOXIRI) compared with infusional fluorouracil, leucovorin, and irinotecan (FOLFIRI) as first-line treatment for metastatic colorectal cancer: the Gruppo Oncologico Nord Ovest. „J Clin Oncol”. 25 (13), s. 1670–1676, maj 2007. DOI: 10.1200/JCO.2006.09.0928. PMID: 17470860.
- ↑ a b J. Souglakos, N. Androulakis, K. Syrigos, A. Polyzos i inni. FOLFOXIRI (folinic acid, 5-fluorouracil, oxaliplatin and irinotecan) vs FOLFIRI (folinic acid, 5-fluorouracil and irinotecan) as first-line treatment in metastatic colorectal cancer (MCC): a multicentre randomised phase III trial from the Hellenic Oncology Research Group (HORG). „Br J Cancer”. 94 (6), s. 798–805, marzec 2006. DOI: 10.1038/sj.bjc.6603011. PMID: 16508637.
- ↑ a b c G. Masi, E. Vasile, F. Loupakis, S. Cupini i inni. Randomized trial of two induction chemotherapy regimens in metastatic colorectal cancer: an updated analysis. „J Natl Cancer Inst”. 103 (1), s. 21–30, styczeń 2011. DOI: 10.1093/jnci/djq456. PMID: 21123833.
- ↑ G. Folprecht, T. Gruenberger, W.O. Bechstein, H.R. Raab i inni. Tumour response and secondary resectability of colorectal liver metastases following neoadjuvant chemotherapy with cetuximab: the CELIM randomised phase 2 trial. „Lancet Oncol”. 11 (1), s. 38–47, styczeń 2010. DOI: 10.1016/S1470-2045(09)70330-4. PMID: 19942479.
- ↑ G. Folprecht, T. Gruenberger, W. Bechstein, H.R. Raab i inni. Survival of patients with initially unresectable colorectal liver metastases treated with FOLFOX/cetuximab or FOLFIRI/cetuximab in a multidisciplinary concept (CELIM study). „Ann Oncol”. 25 (5), s. 1018–1025, maj 2014. DOI: 10.1093/annonc/mdu088. PMID: 24585720.
- ↑ L.C. Ye, T.S. Liu, L. Ren, Y. Wei i inni. Randomized controlled trial of cetuximab plus chemotherapy for patients with KRAS wild-type unresectable colorectal liver-limited metastases. „J Clin Oncol”. 31 (16), s. 1931–1938, czerwiec 2013. DOI: 10.1200/JCO.2012.44.8308. PMID: 23569301.
- ↑ F. Petrelli, S. Barni. Resectability and outcome with anti-EGFR agents in patients with KRAS wild-type colorectal liver-limited metastases: a meta-analysis. „Int J Colorectal Dis”. 27 (8), s. 997–1004, sierpień 2012. DOI: 10.1007/s00384-012-1438-2. PMID: 22358385.
- ↑ C.S. Fuchs, J. Marshall, E. Mitchell, R. Wierzbicki i inni. Randomized, controlled trial of irinotecan plus infusional, bolus, or oral fluoropyrimidines in first-line treatment of metastatic colorectal cancer: results from the BICC-C Study. „J Clin Oncol”. 25 (30), s. 4779–4786, październik 2007. DOI: 10.1200/JCO.2007.11.3357. PMID: 17947725.
- ↑ a b H. Hurwitz, L. Fehrenbacher, W. Novotny, T. Cartwright i inni. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. „N Engl J Med”. 350 (23), s. 2335–2342, czerwiec 2004. DOI: 10.1056/NEJMoa032691. PMID: 15175435.
- ↑ a b L.B. Saltz, S. Clarke, E. Díaz-Rubio, W. Scheithauer i inni. Bevacizumab in combination with oxaliplatin-based chemotherapy as first-line therapy in metastatic colorectal cancer: a randomized phase III study. „J Clin Oncol”. 26 (12), s. 2013–2019, kwiecień 2008. DOI: 10.1200/JCO.2007.14.9930. PMID: 18421054.
- ↑ a b Benson i in. 2015 ↓, s. 65.
- ↑ DeVita, Lawrence i Rosenberg 2008 ↓, s. 1260–1261.
- ↑ a b c Benson i in. 2015 ↓, s. 66.
- ↑ a b Benson i in. 2015 ↓, s. 25–27.
- ↑ Benson i in. 2015 ↓, s. 66–67.
- ↑ a b Benson i in. 2015 ↓, s. 30.
- ↑ a b c Benson i in. 2015 ↓, s. 69.
- ↑ S. Giacchetti, B. Perpoint, R. Zidani, N. Le Bail i inni. Phase III multicenter randomized trial of oxaliplatin added to chronomodulated fluorouracil-leucovorin as first-line treatment of metastatic colorectal cancer. „J Clin Oncol”. 18 (1), s. 136–147, styczeń 2000. PMID: 10623704.
- ↑ a b DeVita, Lawrence i Rosenberg 2008 ↓, s. 1267.
- ↑ T. André, M.A. Bensmaine, C. Louvet, E. François i inni. Multicenter phase II study of bimonthly high-dose leucovorin, fluorouracil infusion, and oxaliplatin for metastatic colorectal cancer resistant to the same leucovorin and fluorouracil regimen. „J Clin Oncol”. 17 (11), s. 3560–3568, listopad 1999. PMID: 10550155.
- ↑ A. de Gramont, A. Figer, M. Seymour, M. Homerin i inni. Leucovorin and fluorouracil with or without oxaliplatin as first-line treatment in advanced colorectal cancer. „J Clin Oncol”. 18 (16), s. 2938–2947, sierpień 2000. PMID: 10944126.
- ↑ B. Nordlinger, H. Sorbye, B. Glimelius, G.J. Poston i inni. Perioperative chemotherapy with FOLFOX4 and surgery versus surgery alone for resectable liver metastases from colorectal cancer (EORTC Intergroup trial 40983): a randomised controlled trial. „Lancet”. 371 (9617), s. 1007–1016, marzec 2008. DOI: 10.1016/S0140-6736(08)60455-9. PMID: 18358928.
- ↑ B. Nordlinger, H. Sorbye, B. Glimelius, G.J. Poston i inni. Perioperative FOLFOX4 chemotherapy and surgery versus surgery alone for resectable liver metastases from colorectal cancer (EORTC 40983): long-term results of a randomised, controlled, phase 3 trial. „Lancet Oncol”. 14 (12), s. 1208–1215, listopad 2013. DOI: 10.1016/S1470-2045(13)70447-9. PMID: 24120480.
- ↑ Benson i in. 2015 ↓, s. 66–68.
- ↑ Tomas Buchler, Tomas Pavlik, Bohuslav Melichar, Zbynek Bortlicek i inni. Bevacizumab with 5-fluorouracil, leucovorin, and oxaliplatin versus bevacizumab with capecitabine and oxaliplatin for metastatic colorectal carcinoma: results of a large registry-based cohort analysis. „BMC Cancer”, 2014. DOI: 10.1186/1471-2407-14-323.
- ↑ Benson i in. 2015 ↓, s. 25.
- ↑ J. Cassidy, S. Clarke, E. Díaz-Rubio, W. Scheithauer i inni. Randomized phase III study of capecitabine plus oxaliplatin compared with fluorouracil/folinic acid plus oxaliplatin as first-line therapy for metastatic colorectal cancer. „J Clin Oncol”. 26 (12), s. 2006–2012, kwiecień 2008. DOI: 10.1200/JCO.2007.14.9898. PMID: 18421053.
- ↑ J. Cassidy, S. Clarke, E. Díaz-Rubio, W. Scheithauer i inni. XELOX vs FOLFOX-4 as first-line therapy for metastatic colorectal cancer: NO16966 updated results. „Br J Cancer”. 105 (1), s. 58–64, czerwiec 2011. DOI: 10.1038/bjc.2011.201. PMID: 21673685.
- ↑ DeVita, Lawrence i Rosenberg 2008 ↓, s. 1271.
- ↑ C. Zhang, J. Wang, H. Gu, D. Zhu i inni. Capecitabine plus oxaliplatin compared with 5-fluorouracil plus oxaliplatin in metastatic colorectal cancer: Meta-analysis of randomized controlled trials. „Oncol Lett”. 3 (4), s. 831–838, kwiecień 2012. DOI: 10.3892/ol.2012.567. PMID: 22741002.
- ↑ Benson i in. 2015 ↓, s. 31.
- ↑ Benson i in. 2015 ↓, s. 26.
- ↑ a b c Benson i in. 2015 ↓, s. 71.
- ↑ C. Tournigand, T. André, E. Achille, G. Lledo i inni. FOLFIRI followed by FOLFOX6 or the reverse sequence in advanced colorectal cancer: a randomized GERCOR study. „J Clin Oncol”. 22 (2), s. 229–237, styczeń 2004. DOI: 10.1200/JCO.2004.05.113. PMID: 14657227.
- ↑ G. Colucci, V. Gebbia, G. Paoletti, F. Giuliani i inni. Phase III randomized trial of FOLFIRI versus FOLFOX4 in the treatment of advanced colorectal cancer: a multicenter study of the Gruppo Oncologico Dell’Italia Meridionale. „J Clin Oncol”. 23 (22), s. 4866–4875, sierpień 2005. DOI: 10.1200/JCO.2005.07.113. PMID: 15939922.
- ↑ Benson i in. 2015 ↓, s. 32.
- ↑ a b Benson i in. 2015 ↓, s. 27.
- ↑ E. Mitry, A.L. Fields, H. Bleiberg, R. Labianca i inni. Adjuvant chemotherapy after potentially curative resection of metastases from colorectal cancer: a pooled analysis of two randomized trials. „J Clin Oncol”. 26 (30), s. 4906–4911, październik 2008. DOI: 10.1200/JCO.2008.17.3781. PMID: 18794541.
- ↑ D. Cunningham, I. Lang, E. Marcuello, V. Lorusso i inni. Bevacizumab plus capecitabine versus capecitabine alone in elderly patients with previously untreated metastatic colorectal cancer (AVEX): an open-label, randomised phase 3 trial. „Lancet Oncol”. 14 (11), s. 1077–1085, październik 2013. DOI: 10.1016/S1470-2045(13)70154-2. PMID: 24028813.
- ↑ P.L. McCormack, S.J. Keam. Bevacizumab: a review of its use in metastatic colorectal cancer. „Drugs”. 68 (4), s. 487–506, 2008. PMID: 18318567.
- ↑ a b c Benson i in. 2015 ↓, s. 73.
- ↑ F. Kabbinavar, H.I. Hurwitz, L. Fehrenbacher, N.J. Meropol i inni. Phase II, randomized trial comparing bevacizumab plus fluorouracil (FU)/leucovorin (LV) with FU/LV alone in patients with metastatic colorectal cancer. „J Clin Oncol”. 21 (1), s. 60–65, styczeń 2003. PMID: 12506171.
- ↑ F.F. Kabbinavar, J. Schulz, M. McCleod, T. Patel i inni. Addition of bevacizumab to bolus fluorouracil and leucovorin in first-line metastatic colorectal cancer: results of a randomized phase II trial. „J Clin Oncol”. 23 (16), s. 3697–3705, czerwiec 2005. DOI: 10.1200/JCO.2005.05.112. PMID: 15738537.
- ↑ F.F. Kabbinavar, J. Hambleton, R.D. Mass, H.I. Hurwitz i inni. Combined analysis of efficacy: the addition of bevacizumab to fluorouracil/leucovorin improves survival for patients with metastatic colorectal cancer. „J Clin Oncol”. 23 (16), s. 3706–3712, czerwiec 2005. DOI: 10.1200/JCO.2005.00.232. PMID: 15867200.
- ↑ Benson i in. 2015 ↓, s. 72.
- ↑ F. Petrelli, K. Borgonovo, M. Cabiddu, M. Ghilardi i inni. FOLFIRI-bevacizumab as first-line chemotherapy in 3500 patients with advanced colorectal cancer: a pooled analysis of 29 published trials. „Clin Colorectal Cancer”. 12 (3), s. 145–151, wrzesień 2013. DOI: 10.1016/j.clcc.2013.04.006. PMID: 23763824.
- ↑ L.T. Macedo, A.B. da Costa Lima, A.D. Sasse. Addition of bevacizumab to first-line chemotherapy in advanced colorectal cancer: a systematic review and meta-analysis, with emphasis on chemotherapy subgroups. „BMC Cancer”. 12, s. 89, 2012. DOI: 10.1186/1471-2407-12-89. PMID: 22414244.
- ↑ J.A. Meyerhardt, L. Li, H.K. Sanoff, W. Carpenter i inni. Effectiveness of bevacizumab with first-line combination chemotherapy for Medicare patients with stage IV colorectal cancer. „J Clin Oncol”. 30 (6), s. 608–615, luty 2012. DOI: 10.1200/JCO.2011.38.9650. PMID: 22253466.
- ↑ H.I. Hurwitz, N.C. Tebbutt, F. Kabbinavar, B.J. Giantonio i inni. Efficacy and safety of bevacizumab in metastatic colorectal cancer: pooled analysis from seven randomized controlled trials. „Oncologist”. 18 (9), s. 1004–1012, 2013. DOI: 10.1634/theoncologist.2013-0107. PMID: 23881988.
- ↑ Y. Cao, A. Tan, F. Gao, L. Liu i inni. A meta-analysis of randomized controlled trials comparing chemotherapy plus bevacizumab with chemotherapy alone in metastatic colorectal cancer. „Int J Colorectal Dis”. 24 (6), s. 677–685, czerwiec 2009. DOI: 10.1007/s00384-009-0655-9. PMID: 19184059.
- ↑ S. Welch, K. Spithoff, R.B. Rumble, J. Maroun. Bevacizumab combined with chemotherapy for patients with advanced colorectal cancer: a systematic review. „Ann Oncol”. 21 (6), s. 1152–1162, czerwiec 2010. DOI: 10.1093/annonc/mdp533. PMID: 19942597.
- ↑ V. Ranpura, S. Hapani, S. Wu. Treatment-related mortality with bevacizumab in cancer patients: a meta-analysis. „JAMA”. 305 (5), s. 487–494, luty 2011. DOI: 10.1001/jama.2011.51. PMID: 21285426.
- ↑ D. Miles, N. Harbeck, B. Escudier, H. Hurwitz i inni. Disease course patterns after discontinuation of bevacizumab: pooled analysis of randomized phase III trials. „J Clin Oncol”. 29 (1), s. 83–88, styczeń 2011. DOI: 10.1200/JCO.2010.30.2794. PMID: 21098326.
- ↑ Benson i in. 2015 ↓, s. 74.
- ↑ Benson i in. 2015 ↓, s. 25–26.
- ↑ a b Benson i in. 2015 ↓, s. 80.
- ↑ a b c d D. Cunningham, Y. Humblet, S. Siena, D. Khayat i inni. Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer. „N Engl J Med”. 351 (4), s. 337–345, lipiec 2004. DOI: 10.1056/NEJMoa033025. PMID: 15269313.
- ↑ a b c E. Van Cutsem, M. Peeters, S. Siena, Y. Humblet i inni. Open-label phase III trial of panitumumab plus best supportive care compared with best supportive care alone in patients with chemotherapy-refractory metastatic colorectal cancer. „J Clin Oncol”. 25 (13), s. 1658–1664, maj 2007. DOI: 10.1200/JCO.2006.08.1620. PMID: 17470858.
- ↑ a b c Benson i in. 2015 ↓, s. 75.
- ↑ J.A. McKay, L.J. Murray, S. Curran, V.G. Ross i inni. Evaluation of the epidermal growth factor receptor (EGFR) in colorectal tumours and lymph node metastases. „Eur J Cancer”. 38 (17), s. 2258–2264, listopad 2002. PMID: 12441262.
- ↑ J.P. Spano, C. Lagorce, D. Atlan, G. Milano i inni. Impact of EGFR expression on colorectal cancer patient prognosis and survival. „Ann Oncol”. 16 (1), s. 102–108, styczeń 2005. DOI: 10.1093/annonc/mdi006. PMID: 15598946.
- ↑ J.R. Hecht, E. Mitchell, M.A. Neubauer, H.A. Burris i inni. Lack of correlation between epidermal growth factor receptor status and response to Panitumumab monotherapy in metastatic colorectal cancer. „Clin Cancer Res”. 16 (7), s. 2205–2213, kwiecień 2010. DOI: 10.1158/1078-0432.CCR-09-2017. PMID: 20332321.
- ↑ K.Y. Chung, J. Shia, N.E. Kemeny, M. Shah i inni. Cetuximab shows activity in colorectal cancer patients with tumors that do not express the epidermal growth factor receptor by immunohistochemistry. „J Clin Oncol”. 23 (9), s. 1803–1810, marzec 2005. DOI: 10.1200/JCO.2005.08.037. PMID: 15677699.
- ↑ H.O. Al-Shamsi, W. Alhazzani, R.A. Wolff. Extended RAS testing in metastatic colorectal cancer-Refining the predictive molecular biomarkers. „J Gastrointest Oncol”. 6 (3), s. 314–321, czerwiec 2015. DOI: 10.3978/j.issn.2078-6891.2015.016. PMID: 26029459.
- ↑ Benson i in. 2015 ↓, s. 77.
- ↑ C. Bokemeyer, I. Bondarenko, A. Makhson, J.T. Hartmann i inni. Fluorouracil, leucovorin, and oxaliplatin with and without cetuximab in the first-line treatment of metastatic colorectal cancer. „J Clin Oncol”. 27 (5), s. 663–671, luty 2009. DOI: 10.1200/JCO.2008.20.8397. PMID: 19114683.
- ↑ T.S. Maughan, R.A. Adams, C.G. Smith, A.M. Meade i inni. Addition of cetuximab to oxaliplatin-based first-line combination chemotherapy for treatment of advanced colorectal cancer: results of the randomised phase 3 MRC COIN trial. „Lancet”. 377 (9783), s. 2103–2114, czerwiec 2011. DOI: 10.1016/S0140-6736(11)60613-2. PMID: 21641636.
- ↑ Julien Taïeb, Tim Maughan, Carsten Bokemeyer, Eric Van Cutsem i inni. Cetuximab combined with infusional 5-fluorouracil/folinic acid (5-FU/FA) and oxaliplatin in metastatic colorectal cancer (mCRC): A pooled analysis of COIN and OPUS study data. „Journal of Clinical Oncology”, 2012.
- ↑ Alan P. Venook, Donna Niedzwiecki, Heinz-Josef Lenz, Federico Innocenti i inni. CALGB/SWOG 80405: Phase III trial of irinotecan/5-FU/leucovorin (FOLFIRI) or oxaliplatin/5-FU/leucovorin (mFOLFOX6) with bevacizumab (BV) or cetuximab (CET) for patients (pts) with KRAS wild-type (wt) untreated metastatic adenocarcinoma of the colon or rectum (MCRC). „Journal of Clinical Oncology”, 2014.
- ↑ E. Van Cutsem, C.H. Köhne, E. Hitre, J. Zaluski i inni. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. „N Engl J Med”. 360 (14), s. 1408–1417, kwiecień 2009. DOI: 10.1056/NEJMoa0805019. PMID: 19339720.
- ↑ I. Láng, C.H. Köhne, G. Folprecht, P. Rougier i inni. Quality of life analysis in patients with KRAS wild-type metastatic colorectal cancer treated first-line with cetuximab plus irinotecan, fluorouracil and leucovorin. „Eur J Cancer”. 49 (2), s. 439–448, styczeń 2013. DOI: 10.1016/j.ejca.2012.08.023. PMID: 23116683.
- ↑ V. Heinemann, L.F. von Weikersthal, T. Decker, A. Kiani i inni. FOLFIRI plus cetuximab versus FOLFIRI plus bevacizumab as first-line treatment for patients with metastatic colorectal cancer (FIRE-3): a randomised, open-label, phase 3 trial. „Lancet Oncol”. 15 (10), s. 1065–1075, wrzesień 2014. DOI: 10.1016/S1470-2045(14)70330-4. PMID: 25088940.
- ↑ J.Y. Douillard, K.S. Oliner, S. Siena, J. Tabernero i inni. Panitumumab-FOLFOX4 treatment and RAS mutations in colorectal cancer. „N Engl J Med”. 369 (11), s. 1023–1034, wrzesień 2013. DOI: 10.1056/NEJMoa1305275. PMID: 24024839.
- ↑ L.S. Schwartzberg, F. Rivera, M. Karthaus, G. Fasola i inni. PEAK: a randomized, multicenter phase II study of panitumumab plus modified fluorouracil, leucovorin, and oxaliplatin (mFOLFOX6) or bevacizumab plus mFOLFOX6 in patients with previously untreated, unresectable, wild-type KRAS exon 2 metastatic colorectal cancer. „J Clin Oncol”. 32 (21), s. 2240–2247, lipiec 2014. DOI: 10.1200/JCO.2013.53.2473. PMID: 24687833.
- ↑ E.P. Mitchell, B. Piperdi, M.E. Lacouture, H. Shearer i inni. The efficacy and safety of panitumumab administered concomitantly with FOLFIRI or Irinotecan in second-line therapy for metastatic colorectal cancer: the secondary analysis from STEPP (Skin Toxicity Evaluation Protocol With Panitumumab) by KRAS status. „Clin Colorectal Cancer”. 10 (4), s. 333–339, grudzień 2011. DOI: 10.1016/j.clcc.2011.06.004. PMID: 22000810.
- ↑ M.T. Seymour, S.R. Brown, S. Richman, G.W. Middleton i inni. Addition of panitumumab to irinotecan: Results of PICCOLO, a randomized controlled trial in advanced colorectal cancer (aCRC). „Journal of Clinical Oncology”, 2011.
- ↑ Alberto F. Sobrero, Marc Peeters, Timothy Jay Price, Yevhen Hotko i inni. Final results from study 181: Randomized phase III study of FOLFIRI with or without panitumumab (pmab) for the treatment of second-line metastatic colorectal cancer (mCRC). „Journal of Clinical Oncology”, 2012.
- ↑ a b Benson i in. 2015 ↓, s. 83.
- ↑ P. Rougier, E. Van Cutsem, E. Bajetta, N. Niederle i inni. Randomised trial of irinotecan versus fluorouracil by continuous infusion after fluorouracil failure in patients with metastatic colorectal cancer. „Lancet”. 352 (9138), s. 1407–1412, październik 1998. DOI: 10.1016/S0140-6736(98)03085-2. PMID: 9807986.
- ↑ G.P. Kim, D.J. Sargent, M.R. Mahoney, K.M. Rowland i inni. Phase III noninferiority trial comparing irinotecan with oxaliplatin, fluorouracil, and leucovorin in patients with advanced colorectal carcinoma previously treated with fluorouracil: N9841. „J Clin Oncol”. 27 (17), s. 2848–2854, czerwiec 2009. DOI: 10.1200/JCO.2008.20.4552. PMID: 19380443.
- ↑ E. Segelov, D. Chan, J. Shapiro, T.J. Price i inni. The role of biological therapy in metastatic colorectal cancer after first-line treatment: a meta-analysis of randomised trials. „Br J Cancer”. 111 (6), s. 1122–1131, wrzesień 2014. DOI: 10.1038/bjc.2014.404. PMID: 25072258.
- ↑ M. Peeters, T.J. Price, A. Cervantes, A.F. Sobrero i inni. Final results from a randomized phase 3 study of FOLFIRI {±} panitumumab for second-line treatment of metastatic colorectal cancer. „Ann Oncol”. 25 (1), s. 107–116, styczeń 2014. DOI: 10.1093/annonc/mdt523. PMID: 24356622.
- ↑ D.J. Jonker, C.J. O’Callaghan, C.S. Karapetis, J.R. Zalcberg i inni. Cetuximab for the treatment of colorectal cancer. „N Engl J Med”. 357 (20), s. 2040–2048, listopad 2007. DOI: 10.1056/NEJMoa071834. PMID: 18003960.
- ↑ Leonard B. Saltz, Neal J. Meropol, Patrick J. Loehrer Sr, Michael N. Needle i inni. Phase II Trial of Cetuximab in Patients With Refractory Colorectal Cancer That Expresses the Epidermal Growth Factor Receptor. „Journal of Clinical Oncology”, 2004. DOI: 10.1200/JCO.2004.10.182.
- ↑ J. Bennouna, J. Sastre, D. Arnold, P. Österlund i inni. Continuation of bevacizumab after first progression in metastatic colorectal cancer (ML18147): a randomised phase 3 trial. „Lancet Oncol”. 14 (1), s. 29–37, styczeń 2013. DOI: 10.1016/S1470-2045(12)70477-1. PMID: 23168366.
- ↑ S. Kubicka, R. Greil, T. André, J. Bennouna i inni. Bevacizumab plus chemotherapy continued beyond first progression in patients with metastatic colorectal cancer previously treated with bevacizumab plus chemotherapy: ML18147 study KRAS subgroup findings. „Ann Oncol”. 24 (9), s. 2342–2349, wrzesień 2013. DOI: 10.1093/annonc/mdt231. PMID: 23852309.
- ↑ Gianluca Masi, Salvatore Salvatore, Fotios Loupakis, Chiara Cremolini i inni. Second-line chemotherapy (CT) with or without bevacizumab (BV) in metastatic colorectal cancer (mCRC) patients (pts) who progressed to a first-line treatment containing BV: Updated results of the phase III „BEBYP” trial by the Gruppo Oncologico Nord Ovest (GONO). „Journal of Clinical Oncology”, 2013.
- ↑ T.H. Cartwright, Y.M. Yim, E. Yu, H. Chung i inni. Survival outcomes of bevacizumab beyond progression in metastatic colorectal cancer patients treated in US community oncology. „Clin Colorectal Cancer”. 11 (4), s. 238–246, grudzień 2012. DOI: 10.1016/j.clcc.2012.05.005. PMID: 22658457.
- ↑ A. Patel, W. Sun. Ziv-aflibercept in metastatic colorectal cancer. „Biologics”. 8, s. 13–25, 2014. DOI: 10.2147/BTT.S39360. PMID: 24368879.
- ↑ E. Van Cutsem, J. Tabernero, R. Lakomy, H. Prenen i inni. Addition of aflibercept to fluorouracil, leucovorin, and irinotecan improves survival in a phase III randomized trial in patients with metastatic colorectal cancer previously treated with an oxaliplatin-based regimen. „J Clin Oncol”. 30 (28), s. 3499–3506, październik 2012. DOI: 10.1200/JCO.2012.42.8201. PMID: 22949147.
- ↑ G. Goel, W. Sun. Ramucirumab, another anti-angiogenic agent for metastatic colorectal cancer in second-line setting-its impact on clinical practice. „J Hematol Oncol”. 8, s. 92, 2015. DOI: 10.1186/s13045-015-0183-8. PMID: 26215324.
- ↑ J. Tabernero, T. Yoshino, A.L. Cohn, R. Obermannova i inni. Ramucirumab versus placebo in combination with second-line FOLFIRI in patients with metastatic colorectal carcinoma that progressed during or after first-line therapy with bevacizumab, oxaliplatin, and a fluoropyrimidine (RAISE): a randomised, double-blind, multicentre, phase 3 study. „Lancet Oncol”. 16 (5), s. 499–508, maj 2015. DOI: 10.1016/S1470-2045(15)70127-0. PMID: 25877855.
- ↑ a b c d e f S.M. Ronnekleiv-Kelly, G.D. Kennedy. Management of stage IV rectal cancer: palliative options. „World J Gastroenterol”. 17 (7), s. 835–847, luty 2011. DOI: 10.3748/wjg.v17.i7.835. PMID: 21412493.
- ↑ a b c d e f g h i j k l m n R. Costi, F. Leonardi, D. Zanoni, V. Violi i inni. Palliative care and end-stage colorectal cancer management: the surgeon meets the oncologist. „World J Gastroenterol”. 20 (24), s. 7602–7621, czerwiec 2014. DOI: 10.3748/wjg.v20.i24.7602. PMID: 24976699.
- ↑ a b c d e f g h i j A. Tuca, E. Guell, E. Martinez-Losada, N. Codorniu. Malignant bowel obstruction in advanced cancer patients: epidemiology, management, and factors influencing spontaneous resolution. „Cancer Manag Res”. 4, s. 159–169, 2012. DOI: 10.2147/CMAR.S29297. PMID: 22904637.
- ↑ M. Winner, S.J. Mooney, D.L. Hershman, D.L. Feingold i inni. Incidence and predictors of bowel obstruction in elderly patients with stage IV colon cancer: a population-based cohort study. „JAMA Surg”. 148 (8), s. 715–722, sierpień 2013. DOI: 10.1001/jamasurg.2013.1. PMID: 23740130.
- ↑ Aljafri A. Majid, Andrew N. Kingsnorth: Advanced Surgical Practice. Cambridge University Press, 2002, s. 202–203. ISBN 978-1-84110-018-0.
- ↑ a b Richard M. Gore, Marc S. Levine: Textbook of Gastrointestinal Radiology. Elsevier Health Sciences, 2008, s. 1104–1105. ISBN 978-0-323-27811-9.
- ↑ M.A. Mohd Suan, W.L. Tan, S.A. Soelar, I. Ismail i inni. Intestinal obstruction: predictor of poor prognosis in colorectal carcinoma?. „Epidemiol Health”. 37, s. e2015017, 2015. DOI: 10.4178/epih/e2015017. PMID: 25868638.
- ↑ a b c d Piotr Liszka-Dalecki, Marek P. Nowacki. Leczenie niedrożności mechanicznej przewodu pokarmowego u pacjentów w zaawansowanym stadium choroby nowotworowej. „Nowa Medycyna”. 4/1999, s. 43–48, 1999.
- ↑ Steven D. Wexner, Neil Stollman: Diseases of the Colon. CRC Press, 2006, s. 24. ISBN 978-1-4200-1857-8.
- ↑ a b c L. Ansaloni, R.E. Andersson, F. Bazzoli, F. Catena i inni. Guidelenines in the management of obstructing cancer of the left colon: consensus conference of the world society of emergency surgery (WSES) and peritoneum and surgery (PnS) society. „World J Emerg Surg”. 5, s. 29, 2010. DOI: 10.1186/1749-7922-5-29. PMID: 21189148.
- ↑ Aljafri A. Majid, Andrew N. Kingsnorth: Advanced Surgical Practice. Cambridge University Press, 2002, s. 203–204. ISBN 978-1-84110-018-0.
- ↑ A.M. Watt, I.G. Faragher, T.T. Griffin, N.A. Rieger i inni. Self-expanding metallic stents for relieving malignant colorectal obstruction: a systematic review. „Ann Surg”. 246 (1), s. 24–30, lipiec 2007. DOI: 10.1097/01.sla.0000261124.72687.72. PMID: 17592286.
- ↑ a b c X.D. Zhao, B.B. Cai, R.S. Cao, R.H. Shi. Palliative treatment for incurable malignant colorectal obstructions: a meta-analysis. „World J Gastroenterol”. 19 (33), s. 5565–5574, wrzesień 2013. DOI: 10.3748/wjg.v19.i33.5565. PMID: 24023502.
- ↑ H. Ptok, F. Marusch, R. Steinert, L. Meyer i inni. Incurable stenosing colorectal carcinoma: endoscopic stent implantation or palliative surgery?. „World J Surg”. 30 (8), s. 1481–1487, sierpień 2006. DOI: 10.1007/s00268-005-0513-z. PMID: 16850152.
- ↑ H. Huhtinen, P. Varpe, J. Karvonen, A. Rantala i inni. Late complications related to palliative stenting in patients with obstructing colorectal cancer. „Minim Invasive Ther Allied Technol”. 22 (6), s. 352–358, grudzień 2013. DOI: 10.3109/13645706.2013.797911. PMID: 23758091.
- ↑ F. Bozzetti, L. Cozzaglio, E. Biganzoli, G. Chiavenna i inni. Quality of life and length of survival in advanced cancer patients on home parenteral nutrition. „Clin Nutr”. 21 (4), s. 281–288, sierpień 2002. PMID: 12135587.
- ↑ B.G. Fan. Parenteral nutrition prolongs the survival of patients associated with malignant gastrointestinal obstruction. „JPEN J Parenter Enteral Nutr”. 31 (6). s. 508–510. PMID: 17947608.
- ↑ a b N. Mandava, S. Kumar, W.F. Pizzi, I.J. Aprile. Perforated colorectal carcinomas. „Am J Surg”. 172 (3), s. 236–238, wrzesień 1996. PMID: 8862074.
- ↑ Ho 2012 ↓, s. 81.
- ↑ a b Ho 2012 ↓, s. 83.
- ↑ Ho 2012 ↓, s. 82.
- ↑ Ho 2012 ↓, s. 83–84.
- ↑ R.K. Portenoy, J. Miransky, H.T. Thaler, J. Hornung i inni. Pain in ambulatory patients with lung or colon cancer. Prevalence, characteristics, and effect. „Cancer”. 70 (6), s. 1616–1624, wrzesień 1992. PMID: 1516015.
- ↑ Szczeklik i Gajewski 2014 ↓, s. 2493–2494.
- ↑ Szczeklik i Gajewski 2014 ↓, s. 2499–2500.
- ↑ a b c d R.A. Swarm, D.L. Anghelescu, C. Benedetti, S. Buga. Adult cancer pain. „J Natl Compr Canc Netw”, s. 47, 2015.
- ↑ R. Plancarte, O.A. de Leon-Casasola, M. El-Helaly, S. Allende i inni. Neurolytic superior hypogastric plexus block for chronic pelvic pain associated with cancer. „Reg Anesth”. 22 (6). s. 562–568. PMID: 9425974.
- ↑ Eduardo Bruera, Irene Higginson, Charles F von Gunten, Tatsuya Morita: Textbook of Palliative Medicine and Supportive Care, Second Edition. CRC Press, 2014, s. 456. ISBN 978-1-4441-3526-8.
- ↑ Sushma Bhatnagar: Freedom From Pain (Sixteenth International Conference Of Indian Association Of Palliative Care). I. K. International Pvt Ltd, 2009, s. 23. ISBN 978-93-80026-18-3.
- ↑ A.M. Nitschke, C.E. Ray. Percutaneous neurolytic celiac plexus block. „Semin Intervent Radiol”. 30 (3), s. 318–321, wrzesień 2013. DOI: 10.1055/s-0033-1353485. PMID: 24436554.
- ↑ D.P. Seamans, G.Y. Wong, J.L. Wilson. Interventional pain therapy for intractable abdominal cancer pain. „J Clin Oncol”. 21 (9 Suppl), s. 92s-94s, maj 2003. DOI: 10.1200/JCO.2003.01.184. PMID: 12743207.
- ↑ Howard S. Smith: Current Therapy in Pain. Elsevier Health Sciences, 2009, s. 223–226. ISBN 978-1-4160-4836-7.
- ↑ Manohar Sharma, Karen Simpson, Michael Bennett, Sanjeeva Gupta: Practical Management of Complex Cancer Pain. OUP Oxford, 2014, s. 163. ISBN 978-0-19-163765-0.
- ↑ Nalini Vadivelu, Richard D. Urman, Roberta L. Hines: Essentials of Pain Management. Springer, 2011, s. 226. ISBN 978-0-387-87579-8.
- ↑ C. Granda-Cameron, D. DeMille, M.P. Lynch, C. Huntzinger i inni. An interdisciplinary approach to manage cancer cachexia. „Clin J Oncol Nurs”. 14 (1), s. 72–80, luty 2010. DOI: 10.1188/10.CJON.72-80. PMID: 20118029.
- ↑ W.D. Dewys, C. Begg, P.T. Lavin, P.R. Band i inni. Prognostic effect of weight loss prior to chemotherapy in cancer patients. Eastern Cooperative Oncology Group. „Am J Med”. 69 (4), s. 491–497, październik 1980. PMID: 7424938.
- ↑ J.N. Gordon, S.R. Green, P.M. Goggin. Cancer cachexia. „QJM”. 98 (11), s. 779–788, listopad 2005. DOI: 10.1093/qjmed/hci127. PMID: 16214835.
- ↑ R. Dhanapal, T. Saraswathi, R.N. Govind. Cancer cachexia. „J Oral Maxillofac Pathol”. 15 (3), s. 257–260, wrzesień 2011. DOI: 10.4103/0973-029X.86670. PMID: 22144825.
- ↑ N.B. Kumar, A. Kazi, T. Smith, T. Crocker i inni. Cancer cachexia: traditional therapies and novel molecular mechanism-based approaches to treatment. „Curr Treat Options Oncol”. 11 (3–4), s. 107–117, grudzień 2010. DOI: 10.1007/s11864-010-0127-z. PMID: 21128029.
- ↑ a b C.L. Donohoe, A.M. Ryan, J.V. Reynolds. Cancer cachexia: mechanisms and clinical implications. „Gastroenterol Res Pract”. 2011, s. 601434, 2011. DOI: 10.1155/2011/601434. PMID: 21760776.
- ↑ a b T. Aoyagi, K.P. Terracina, A. Raza, H. Matsubara i inni. Cancer cachexia, mechanism and treatment. „World J Gastrointest Oncol”. 7 (4), s. 17–29, kwiecień 2015. DOI: 10.4251/wjgo.v7.i4.17. PMID: 25897346.
- ↑ A. Inui. Cancer anorexia-cachexia syndrome: current issues in research and management. „CA Cancer J Clin”. 52 (2). s. 72–91. PMID: 11929007.
- ↑ D.C. McMillan, S.J. Wigmore, K.C. Fearon, P. O’Gorman i inni. A prospective randomized study of megestrol acetate and ibuprofen in gastrointestinal cancer patients with weight loss. „Br J Cancer”. 79 (3–4), s. 495–500, luty 1999. DOI: 10.1038/sj.bjc.6690077. PMID: 10027319.
- ↑ K. Lundholm, J. Gelin, A. Hyltander, C. Lönnroth i inni. Anti-inflammatory treatment may prolong survival in undernourished patients with metastatic solid tumors. „Cancer Res”. 54 (21), s. 5602–5606, listopad 1994. PMID: 7923204.
- ↑ L.C. Cerchietti, A.H. Navigante, M.A. Castro. Effects of eicosapentaenoic and docosahexaenoic n-3 fatty acids from fish oil and preferential Cox-2 inhibition on systemic syndromes in patients with advanced lung cancer. „Nutr Cancer”. 59 (1), s. 14–20, 2007. DOI: 10.1080/01635580701365068. PMID: 17927497.
- ↑ C. Gridelli, C. Gallo, A. Ceribelli, V. Gebbia i inni. Factorial phase III randomised trial of rofecoxib and prolonged constant infusion of gemcitabine in advanced non-small-cell lung cancer: the GEmcitabine-COxib in NSCLC (GECO) study. „Lancet Oncol”. 8 (6), s. 500–512, czerwiec 2007. DOI: 10.1016/S1470-2045(07)70146-8. PMID: 17513173.
- ↑ J. Reid, C.M. Hughes, L.J. Murray, C. Parsons i inni. Non-steroidal anti-inflammatory drugs for the treatment of cancer cachexia: a systematic review. „Palliat Med”. 27 (4), s. 295–303, kwiecień 2013. DOI: 10.1177/0269216312441382. PMID: 22450159.
- ↑ N. Nagaya, M. Kojima, K. Kangawa. Ghrelin, a novel growth hormone-releasing peptide, in the treatment of cardiopulmonary-associated cachexia. „Intern Med”. 45 (3), s. 127–134, 2006. PMID: 16508225.
- ↑ M.D. DeBoer, D.L. Marks. Therapy insight: Use of melanocortin antagonists in the treatment of cachexia in chronic disease. „Nat Clin Pract Endocrinol Metab”. 2 (8), s. 459–466, sierpień 2006. DOI: 10.1038/ncpendmet0221. PMID: 16932335.
- ↑ J.M. Scarlett, D.L. Marks. The use of melanocortin antagonists in cachexia of chronic disease. „Expert Opin Investig Drugs”. 14 (10), s. 1233–1239, październik 2005. DOI: 10.1517/13543784.14.10.1233. PMID: 16185165.
- ↑ S. Busquets, M.T. Figueras, G. Fuster, V. Almendro i inni. Anticachectic effects of formoterol: a drug for potential treatment of muscle wasting. „Cancer Res”. 64 (18), s. 6725–6731, wrzesień 2004. DOI: 10.1158/0008-5472.CAN-04-0425. PMID: 15374990.
- ↑ J.N. Gordon, T.M. Trebble, R.D. Ellis, H.D. Duncan i inni. Thalidomide in the treatment of cancer cachexia: a randomised placebo controlled trial. „Gut”. 54 (4), s. 540–545, kwiecień 2005. DOI: 10.1136/gut.2004.047563. PMID: 15753541.
- ↑ a b Stephen Hauser: Mayo Clinic Gastroenterology and Hepatology Board Review. Oxford University Press, 2014, s. 187. ISBN 978-0-19-937335-2.
- ↑ a b Karen E. Kim: Early Detection and Prevention of Colorectal Cancer. SLACK Incorporated, 2009, s. 4. ISBN 978-1-55642-837-1.
- ↑ Health at a Glance 2015 OECD Indicators. Organization for Economic Cooperation and Development, 2015, s. 154. DOI: 10.1787/health_glance-2015-en. ISBN 978-92-64-23257-0.
- ↑ What are the survival rates for colorectal cancer by stage?. [dostęp 2015-12-02]. [zarchiwizowane z tego adresu (2015-12-02)].
- ↑ a b c d e D. J. Th. Wagener: The History of Oncology. Houten: Springer, 2009, s. 93–94. ISBN 978-90-313-6143-4.
- ↑ Stephen M. Cohn, Steven T. Brower: Surgery: Evidence-Based Practice. PMPH-USA, 2012, s. 273. ISBN 978-1-60795-109-4.
- ↑ a b c Welch, Ottinger i Welch 2012 ↓, s. 4.
- ↑ a b c d Milica Nestorovic i inni, One hundred years of miles’ operation – what has changed?
- ↑ a b F.G. Campos, A. Habr-Gama, S.C. Nahas, R.O. Perez i inni. Abdominoperineal excision: evolution of a centenary operation. „Dis Colon Rectum”. 55 (8), s. 844–853, sierpień 2012. DOI: 10.1097/DCR.0b013e31825ab0f7. PMID: 22810469.
- ↑ W.C. Hargrove, M.H. Gertner, W.T. Fitts, P. Kraske. The Kraske operation for carcinoma of the rectum. „Surg Gynecol Obstet”. 148 (6), s. 931–933, czerwiec 1979. PMID: 377528.
- ↑ a b Brian G. Czito, Christopher G. Willett: Rectal Cancer: International Perspectives on Multimodality Management. Springer Science & Business Media, 2010, s. 54. ISBN 978-1-60761-567-5.
- ↑ M.W. Büchler, R.J. Heald, B. Ulrich, J. Weitz: Rectal Cancer Treatment. Springer Science & Business Media, 2006, s. 122. ISBN 978-3-540-27449-0.
- ↑ R.J. Heald, E.M. Husband, R.D. Ryall. The mesorectum in rectal cancer surgery--the clue to pelvic recurrence?. „Br J Surg”. 69 (10), s. 613–616, październik 1982. PMID: 6751457.
- ↑ J. Yoo. Laparoscopic colorectal surgery. „Perm J”. 12 (1), s. 27–31, 2008. PMID: 21369509.
- ↑ J.R. Bertino. Chemotherapy of colorectal cancer: history and new themes. „Semin Oncol”. 24 (5 Suppl 18), s. S18-3-S18-7, październik 1997. PMID: 9420015.
- ↑ P. Seifert, L.H. Baker, M.L. Reed, V.K. Vaitkevicius. Comparison of continuously infused 5-fluorouracil with bolus injection in treatment of patients with colorectal adenocarcinoma. „Cancer”. 36 (1), s. 123–128, lipiec 1975. PMID: 1203840.
- ↑ J. Lokich. Infusional 5-FU: historical evolution, rationale, and clinical experience. „Oncology (Williston Park)”. 12 (10 Suppl 7), s. 19–22, październik 1998. PMID: 9830620.
- ↑ B.G. Czito, C.G. Willett. Thirty years of rectal cancer research: a brief history. „Oncology (Williston Park)”. 22 (12), s. 1441–1442, 1444, listopad 2008. PMID: 19322951.
- ↑ a b B. Gustavsson, G. Carlsson, D. Machover, N. Petrelli i inni. A review of the evolution of systemic chemotherapy in the management of colorectal cancer. „Clin Colorectal Cancer”. 14 (1), s. 1–10, marzec 2015. DOI: 10.1016/j.clcc.2014.11.002. PMID: 25579803.
- ↑ E. Mini, F. Trave, Y.M. Rustum, J.R. Bertino. Enhancement of the antitumor effects of 5-fluorouracil by folinic acid. „Pharmacol Ther”. 47 (1), s. 1–19, 1990. PMID: 2195551.
- ↑ L.B. Saltz, J.V. Cox, C. Blanke, L.S. Rosen i inni. Irinotecan plus fluorouracil and leucovorin for metastatic colorectal cancer. Irinotecan Study Group. „N Engl J Med”. 343 (13), s. 905–914, wrzesień 2000. DOI: 10.1056/NEJM200009283431302. PMID: 11006366.
- ↑ R.M. Goldberg, D.J. Sargent, R.F. Morton, C.S. Fuchs i inni. A randomized controlled trial of fluorouracil plus leucovorin, irinotecan, and oxaliplatin combinations in patients with previously untreated metastatic colorectal cancer. „J Clin Oncol”. 22 (1), s. 23–30, styczeń 2004. DOI: 10.1200/JCO.2004.09.046. PMID: 14665611.
- ↑ a b c d Jack E. Moulton: Tumors in Domestic Animals. University of California Press, 1978, s. 263–266. ISBN 978-0-520-02386-4.
- ↑ Ivan Bubanovic: Origin of Anti-Tumor Immunity Failure in Mammals. Springer, 2004, s. 197. ISBN 978-0-306-48630-2.
- ↑ Gerald Hoff: Diseases of Amphibians and Reptiles. Springer Science & Business Media, 1984, s. 548. ISBN 978-1-4615-9393-5.
- ↑ a b c R.L. Johnson, J.C. Fleet. Animal models of colorectal cancer. „Cancer Metastasis Rev”. 32 (1–2), s. 39–61, czerwiec 2013. DOI: 10.1007/s10555-012-9404-6. PMID: 23076650.
- ↑ A. Velcich, W. Yang, J. Heyer, A. Fragale i inni. Colorectal cancer in mice genetically deficient in the mucin Muc2. „Science”. 295 (5560), s. 1726–1729, marzec 2002. DOI: 10.1126/science.1069094. PMID: 11872843.
- ↑ a b S.D. Taylor, N. Pusterla, B. Vaughan, M.B. Whitcomb i inni. Intestinal neoplasia in horses. „J Vet Intern Med”. 20 (6). s. 1429–1436. PMID: 17186861.
- ↑ a b c d Stephen J. Withrow, David M. Vail: Withrow and MacEwen’s Small Animal Clinical Oncology. Elsevier Health Sciences, 2007, s. 491–500. ISBN 978-0-7216-0558-6.
- ↑ a b c d e Rafał Sapierzyński. Nowotwory układu pokarmowego u psów i kotów. Część II. Nowotwory przełyku, żołądka i jelit. „Życie Weterynaryjne”, 2006. [zarchiwizowane z adresu 2016-03-05].
- ↑ a b Stephen J. Withrow, David M. Vail, Rodney Page: Withrow and MacEwen’s Small Animal Clinical Oncology. Elsevier Health Sciences, 2013, s. 420. ISBN 978-0-323-24197-7.
Bibliografia
- H. Randolph Bailey, Richard P. Billingham, Michael J. Stamos, Michael J. Snyder: Colorectal Surgery. Elsevier Health Sciences, 2012. ISBN 978-1-4557-3770-3.
- A.B. Benson, A.P. Venook, T. Bekaii-Saab, E. Chan i inni. Colon cancer, version 3.2015. „J Natl Compr Canc Netw”, 2015.
- Jim Cassidy, Patrick Johnston, Eric Van Cutsem: Colorectal Cancer. CRC Press, 2006. ISBN 978-1-4200-1630-7.
- Vincent T. DeVita, Theodore S. Lawrence, Steven A. Rosenberg: Devita, Hellman & Rosenberg’s Cancer: Principles & Practice of Oncology. Wyd. 8. Lippincott Williams & Wilkins, 2008. ISBN 978-0-7817-7207-5.
- Peter C. Enzinger, Moon J. Fenton, Charles S. Fuchs, Jean L. Grem i inni. Rectal Cancer, Version 3.2015. „J Natl Compr Canc Netw”, 2015.
- B. Glimelius, E. Tiret, A. Cervantes, D. Arnold. Rectal cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. „Ann Oncol”. 24 Suppl 6, s. vi81–vi88, październik 2013. DOI: 10.1093/annonc/mdt240. PMID: 24078665.
- S.R. Hamilton, L.A. Aaltonen, Pathology and Genetics of Tumours of the Digestive System, Lyon: IARC Press, 2000, ISBN 92-832-2410-8 [dostęp 2015-12-25] [zarchiwizowane z adresu 2018-07-28].
- Yik-Hong Ho: Contemporary Issues in Colorectal Surgical Practice. INTECH, 2012. ISBN 978-953-51-0257-1.
- David Kelsen: Principles and Practice of Gastrointestinal Oncology. Lippincott Williams & Wilkins, 2008. ISBN 978-0-7817-7617-2.
- Radzisław Kordek: Onkologia. Podręcznik dla studentów i lekarzy. Gdańsk: VIA MEDICA, 2007. ISBN 978-83-7555-016-0.
- Maciej Krzakowski, Piotr Potemski, Krzysztof Warzocha, Pior Wysocki: Onkologia kliniczna. T. II. Via Medica, 2015. ISBN 978-83-7599-796-5.
- Vinay Kumar, Ramzi S. Cotran, Stanley L. Robins: Robins Patologia. Wrocław: Elsevier Urban & Partner, 2005, s. 232–232. ISBN 83-89581-92-2.
- R. Labianca, B. Nordlinger, G.D. Beretta, S. Mosconi i inni. Early colon cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. „Ann Oncol”. 24 Suppl 6, s. vi64-72, październik 2013. DOI: 10.1093/annonc/mdt354. PMID: 24078664.
- Bernard Levin, Paul Rozen, Stephen J. Spann, Graeme P. Young: Colorectal Cancer in Clinical Practice: Prevention, Early Detection and Management. CRC Press, 2005. ISBN 978-1-84184-455-8.
 Otto S. Lin. Acquired risk factors for colorectal cancer. „Methods in Molecular Biology”. 472, s. 361–372, 2009. DOI: 10.1007/978-1-60327-492-0_16. PMID: 19107442.
Otto S. Lin. Acquired risk factors for colorectal cancer. „Methods in Molecular Biology”. 472, s. 361–372, 2009. DOI: 10.1007/978-1-60327-492-0_16. PMID: 19107442.- Robert D. Odze Robert D. Odze, John R. Goldblum John R. Goldblum: Surgical Pathology of the GI Tract, Liver, Biliary Tract and Pancreas. Elsevier Health Sciences, 2009. ISBN 978-1-4377-1959-8.
- Daniel K. Podolsky, Michael Camilleri, J. Gregory Fitz, Anthony N. Kalloo, Fergus Shanahan, Timothy C. Wang: Yamada’s Textbook of Gastroenterology. John Wiley & Sons, 2015. ISBN 978-1-118-51215-9.
- Pat Price, Karol Sikora: Treatment of cancer. CRC Press, 2014. ISBN 978-1-4822-1494-9.
- Elaine Swan: Colorectal Cancer. John Wiley & Sons, 2006. ISBN 978-0-470-03168-1.
- Andrzej Szczeklik, Piotr Gajewski: Interna Szczeklika 2014. Kraków: Medycyna Praktyczna, 2014. ISBN 978-83-7430-405-4.
- E. Van Cutsem, A. Cervantes, B. Nordlinger, D. Arnold i inni. Metastatic colorectal cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. „Ann Oncol”. 25 Suppl 3, s. iii1–9, wrzesień 2014. DOI: 10.1093/annonc/mdu260. PMID: 25190710.
- Claude E. Welch, Leslie W. Ottinger, John P. Welch: Manual of Lower Gastrointestinal Surgery. Springer Science & Business Media, 2012. ISBN 978-1-4612-6012-7.
- Christopher G. Willett: Cancer of the Lower Gastrointestinal Tract. T. 1. PMPH-USA, 2001, s. 82. ISBN 9781550091106.
![]() Przeczytaj ostrzeżenie dotyczące informacji medycznych i pokrewnych zamieszczonych w Wikipedii.
Przeczytaj ostrzeżenie dotyczące informacji medycznych i pokrewnych zamieszczonych w Wikipedii.
Media użyte na tej stronie

The Star of Life, medical symbol used on some ambulances.
Star of Life was designed/created by a National Highway Traffic Safety Administration (US Gov) employee and is thus in the public domain.(c) Emmanuelm at en.wikipedia, CC BY 3.0
en:Colorectal cancer, gross appearance of an opened en:colectomy specimen.
Autor: Nephron, Licencja: CC BY-SA 3.0
Low micrograph of a colorectal tubular adenoma without high grade dysplasia. H&E stain.
The lesional tissue, i.e. dysplastic epithelium, is seen on the left of the image and characterized by:
- Nuclear hyperchromatism (i.e. dark purple nuclei) - that extends to the surface,
- Nuclear crowding (i.e. nuclei are bunched-up),
- Elliptical/cigar-shaped nuclei and loss of goblet cells/reduction in the number of goblet cells.
Normal colonic type epithelium is seen on the right of the image and characterized by small round nuclei and abundant goblet cells.
Related images
The same case:

Low mag.

Intermed. mag.
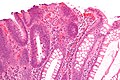
High mag.



Another case:

High mag.

Autor: Gilo1969, Licencja: CC BY-SA 3.0
Endoscopy snare used for upper GI and colonic polypectomy
Autor:
- Fæ
- Cancer_Research_UK_uploader
- derivative work: M1llx (zgłoś błąd)
Zakres resekcji w guzie poprzecznicy.
Autor: Nephron, Licencja: CC BY-SA 3.0
Micrograph showing tumour-infiltrating lymphocytes in colorectal carcinoma. H&E stain.
Tumour-infiltrating lymphocytes (TILs) are suggestive of microsatellite instability (MSI), and may be seen in Lynch syndrome.
Related images

TILs - high mag.

TILs - very high mag.
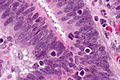
TILs - extremely high mag.




TILs - high mag.

TILs - very high mag.


Autor:
- Fæ
- Cancer_Research_UK_uploader
- derivative work: M1llx (zgłoś błąd)
Zakres resekcji w guzie kątnicy i wstępnicy.
Autor:
- Fæ
- Cancer_Research_UK_uploader
- derivative work: M1llx (zgłoś błąd)
Zakres resekcji w guzie zstępnicy.
Autor: 邱鈺鋒, Licencja: CC BY-SA 4.0
大腸癌經常經由肝門靜脈轉移至肝臟,在肝臟造成極大的傷害,最後嚴重威脅罹癌病患的生命。
Autor: Kazunari Mado, Yukimoto Ishii, Takero Mazaki, Masaya Ushio, Hideki Masuda and Tadatoshi Takayama, Licencja: CC BY-SA 2.0
A) 99mTc-HMDP bone scintigraphy showing many lesions with accumulation in the left ribs, which were diagnosed as multiple costal metastases. B) After chemotherapy with S-1 and CPT-11, the costal metastases have resolved..
Autor: Nephron, Licencja: CC BY-SA 3.0
Micrograph of a traditional serrated adenoma. H&E stain.
Related images

TSA - low mag.

TSA - intermed. mag.
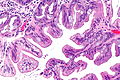
TSA - high mag.

TSA - very high mag.




Six bottles of different types of cancer drugs over a graded blue to white background. Clockwise from center: Blenoxane (bleomycin), Oncovin (vincristine), DTIC-Dome (dacarbazine), Cytoxan (cyclophosphamide), Adriamycin (doxorubicin), and VePesid (etoposide).
Autor: Nephron, Licencja: CC BY-SA 3.0
Micrograph of a colorectal adenocarcinoma metastasis to a lymph node, also lymph node metastasis. H&E stain.
The cancer (forming glands) is seen at the centre-top. Adipose tissue is present on the upper right.
See also
- Image:Breast carcinoma in a lymph node.jpg - met to a lymph node in breast carcinoma.
- Image:Lymph node with papillary thyroid carcinoma.jpg - met to a lymph node in papillary thyroid carcinoma.
Autor: Nephron, Licencja: CC BY-SA 3.0
Micrograph of a colorectal adenocarcinoma. H&E stain.
Related images

CRC - low mag.

CRC - intermed. mag.

CRC - high mag.



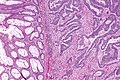
CRC - low mag.

CRC - intermed. mag.

CRC - high mag.



- Image title: Porterhouse steaks grilling barbecue grills meat
- Image from Public domain images website, http://www.public-domain-image.com/full-image/food-and-drink-public-domain-images-pictures/barbecue-grilling-food-public-domain-images-pictures/porterhouse-steaks-grilling-barbecue-grills-meat.jpg.html
This is the same case presented in Image:Tubulovillous Polyp of the Colon 1.jpg
The polyp is shown in longitudinal section. Resembling congealed beef buillon, the hemorrhagic submucosa of the stalk and colon wall proper is set off strikingly from the pale ribbon of mucosa that covers it.
Both photos were taken with a Minolta X-370 with 100mm Rokkor bellows lens, illuminated by 4 photofloods, on Kodak Elite ISO 100 daylight slide film, with 80B blue filter to compensate for tungsten color temperature.
Photograph by Ed Uthman, MD. Public domain. Posted 28 May 00ID#: 62 Description: Gross pathology of adenocarcinoma, colon
Exophytic adenocarcinoma of colon. Surgical specimen.
Content Providers(s): CDC/ Dr. Edwin P. Ewing, Jr.
Creation Date: 1974
Copyright Restrictions: None - This image is in the public domain and thus free of any copyright restrictions. As a matter of courtesy we request that the content provider be credited and notified in any public or private usage of this image.Autor:
- Fæ
- Cancer_Research_UK_uploader
- derivative work: M1llx (zgłoś błąd)
Zaawansowanie guza pierwotnego (cecha T) według klasyfikacji TNM.
Autor: Gilo1969, Licencja: CC BY-SA 3.0
Identification and removal of a pedunculated colonic polyp at colonoscopy
Fentanyl patch packages from several german generic drug manufacturers (from left to right: 1A Pharma, Winthrop, TAD Pharma, ratiopharm, Hexal).
Autor: Yale Rosen from USA, Licencja: CC BY-SA 2.0
Metastatic colonic adenocarcinoma
Autor: Olek Remesz (wiki-pl: Orem, commons: Orem), Licencja: CC BY-SA 2.5
Schemat przewodu pokarmowego, z zaznaczonymi częściami jelita grubego.
Autor: Autor nie został podany w rozpoznawalny automatycznie sposób. Założono, że to Sjoehest (w oparciu o szablon praw autorskich)., Licencja: CC-BY-SA-3.0
de:
- Bildbeschreibung: Während einer Koloskopie aufgenommenes Foto eines Karzinomes des Colon ascendens. Mit dem Instrument am unteren Bildrand wird eine Biopsie entnommen.
- Fotograf: Selbst angefertigt von de:Benutzer:Doktor silke
- Datum: 27.09.2005
- andere Versionen: nein
en:
- Description: during a colonoskopy made image of a carcinoma of the colon ascendens with the tool, which can be seen at the lower border is used for a biopsy
- Photographer: Doktor silke
- Date: 2005/09/27
- Older versions: none
Autor: Department of Pathology, Calicut Medical College, Licencja: CC BY-SA 4.0
Cut open section of colectomy specimen showing hundreds of small polyps throughout the entire colon
ID#: 64 Description: Gross pathology of adenocarcinoma, colon
"Napkin-ring" configuration of colonic adenocarcinoma. Surgical specimen.
Content Providers(s): CDC/ Dr. Edwin P. Ewing, Jr.
Creation Date: 1974
Copyright Restrictions: None - This image is in the public domain and thus free of any copyright restrictions. As a matter of courtesy we request that the content provider be credited and notified in any public or private usage of this image.Autor: RobLekarzMD, Licencja: CC BY-SA 4.0
Uszypułowany polip z głową o średnicy ok 40 mm.
Autor: James Heilman, MD, Licencja: CC BY-SA 3.0
A positive fecal occult blood test.
Fruit on display at La Boqueria market in Barcelona.
Autor: Anpol42, Licencja: CC BY-SA 4.0
Intraoperativ view (laparoscopic operation) of malignant tumor of sigma
Autor:
- Fæ
- Cancer_Research_UK_uploader
- derivative work: M1llx (zgłoś błąd)
Zakres resekcji w guzie esicy.
Autor:
-
Bernstein0275
- derivative work: M1llx (zgłoś błąd)
Wzdłużnie rozcięty fragment jelita, z widocznymi czterema polipami i rakiem. Poniżej dodany diagram z zaznaczonym prawdopodobnym obszarem zmiany przedrakowej (obszar tkanki, który poprzedza i predysponuje do rozwoju raka). Schemat wskazuje kolejne mutacje klonalne, które były prekursorami guzów.
Autor: Ed Uthman from Houston, TX, USA, Licencja: CC BY 2.0
Note that the cancer (left) meets normal mucosa (right) abruptly, with no evidence of a pre-existing adenoma.

(c) Samir z angielskojęzycznej Wikipedii, CC-BY-SA-3.0
Image of en:familial adenomatous polyposis as seen on en:sigmoidoscopy. Released into public domain on permission of patient. -- Samir धर्म 06:07, 10 August 2006 (UTC)
en:Category:Endoscopic imagesAutor: The Juan Rosai Collection, Licencja: CC0
Microscopically the diagnosis of a villous adenoma is easily confirmed by observation of the enlarged glands containing enlarged cells with hypochromatic nuclei that are stratified and have a typical picket fence appearance and an obvious villous architecture.

(c) Emmanuelm z angielskiej Wikipedii, CC BY 3.0
Gross appearance of an opened colectomy specimen containing an invasive colorectal carcinoma and two adenomatous polyps.
Autor: Ed Uthman from Houston, TX, USA, Licencja: CC BY 2.0
In an adenocarcinoma of the colon of a man who presented with widely metastatic disease and no localizing symptoms.
Autor:
- Originally from {Molecular cell biology. Lodish, Harvey 5 ed: - New York : W. H. Freeman and Co., 2003, 973 s. b ill. ISBN: 0-7167-4366-3. Libris: 8926100. The Hallmarks of Cancer, Douglas Hanahan and Robert A. Weinberg, Cell, Vol. 100, 57–70, January 7, 2000. http://www.weizmann.ac.il/home/fedomany/Bioinfo05/lecture6_Hanahan.pdf
- Roadnottaken z angielskiej Wikipedii
- User:Cybertory
- derivative work: M1llx (zgłoś błąd)
Uproszczony schemat ważnych szlaków sygnałowych.
--> = aktywacja
--| = inhibicjaAutor: Ed Uthman from Houston, TX, USA, Licencja: CC BY 2.0
This large villous adenoma carpets the cecum of a right colectomy specimen. The fixed specimen is immersed in tapwater to display the delicate filigree of the villous surface pattern.
To view another (better) presentation of this image, check out the version on the Website of Objective Pathology.
Autor:
- Fæ
- Cancer_Research_UK_uploader
- derivative work: M1llx (zgłoś błąd)
Diagram przedstawiający miejscową resekcję wczesnego stadium raka jelita grubego.
Autor: Anpol42, Licencja: CC BY-SA 4.0
Laparoskopowa resekcja esicy przy pomocy staplera liniowego.
Autor: Cbalentine, Licencja: CC BY-SA 4.0
Test immunochemiczny w kale do wykrywania krwi utajonej w kale, powszechnie stosowany w badaniach przesiewowych w kierunku raka jelita grubego.
PET/CT of a staging exam of colon carcinoma. Besides the primary tumor a lot of lesions can be seen. On cursor position: lung nodule. Imaging parameters: Patient: 69kg, 186cm. Scanned 45 minutes after injection. Injected dose: 300mBq FDG, total acquisition time: 6 minutes. Device: GE PET/CT Discovery 600
Autor: Yale Rosen from USA, Licencja: CC BY-SA 2.0
Metastatic colonic adenocarcinoma
Autor: PanSG, Licencja: CC BY-SA 4.0
Rak okrężnicy - wykres zachorowalnośći
Autor: Ed Uthman from Houston, TX, USA, Licencja: CC BY 2.0
Adenocarcinoma of the Sigmoid Colon (en face, closeup)
Autor: Anpol42, Licencja: CC BY-SA 4.0
A - Zakres resekcji esicy (zaznaczony szaro); B - zamknięty kikut odbytnicy; C - Kolostomia (odbyt sztuczny)
Autor: Patho, Licencja: CC BY-SA 3.0
tumor invasion into vein in a case of colorectal cancer.
Autor: Nephron, Licencja: CC BY-SA 3.0
Very low magnification micrograph of a mucinous adenocarcinoma of the colon, a type of colon cancer. H&E stain.
Related images

Very low mag.

Low mag.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Autor: Ed Uthman from Houston, TX, USA, Licencja: CC BY 2.0
The patient, a middle-aged woman, presented with colon perforation and paracolonic abscess. Diagnosis was made at laparotomy.
Autor: liz west from Boxborough, MA, Licencja: CC BY 2.0
First look at the radiation machine, which doesn't look all that threatening. It makes different noises from the other machines, however.
Autor: Ed Uthman from Houston, TX, USA, Licencja: CC BY 2.0
Adenocarcinoma of the Sigmoid Colon (longitudinal section)'
This long shot of a longitudinal section shows the tumor at the center, bounded on either side by submucosa that is distended by tattoo pigment.Autor:
- Fæ
- Cancer_Research_UK_uploader
- derivative work: M1llx (zgłoś błąd)
Zakres resekcji w guzie odbytnicy z wyłonieniem stałej stomii.
Autor: Ed Uthman from Houston, TX, USA, Licencja: CC BY 2.0
The is the same subject shown in the previous image, except that it is immersed in alcohol. Although water can also be used, alcohol is easier to work with. Since tissue has a higher specific gravity than alcohol, the subject won't float to the top of the liquid. Also, tiny bits of tissue, which would float around in water and cloud the image, sink right to the bottom and stay out of the way. This makes alcohol seem "clearer" than water.
Vincenz Czerny (1842-1916) war ein deutscher Chirurg und Krebsforscher.
(c) Lokal_Profil, CC BY-SA 2.5
Age-standardised death rates from Colon and rectum cancers by country (per 100,000 inhabitants).
The Tuscan General Alessandro del Borro (?)