Zespół sercowo-twarzowo-skórny
 7,5-letni chłopiec ze zdiagnozowanym zespołem sercowo-twarzowo-skórnym (widoczne są grube rysy twarzy, kręcone suche włosy, rzadkie brwi, szeroki czubek nosa z antewersją nozdrzy)[1] | |
| ICD-10 | Q87.8 |
|---|---|
| DiseasesDB | |
| OMIM | |
| MeSH | |
Zespół sercowo-twarzowo-skórny (ang. cardiofaciocutaneous syndrome, CFC syndrome) – genetycznie uwarunkowany zespół wad wrodzonych dziedziczony autosomalnie dominująco. Charakteryzuje się niedoborem wzrostu, typowymi cechami dysmorficznymi w twarzy, wadami serca, zmianami skórnymi oraz niepełnosprawnością intelektualną.
Historia
Zespół został opisany po raz pierwszy w 1986 roku przez amerykańskich lekarzy Jamesa Reynoldsa, Giovanniego Neriego, Jurgena Herrmanna, Bruce’a Blumberga, Jamesa Coldwella, Paula Milesa, Johna Opitza i Johna Care’a[2].
Etiologia
Choroba spowodowana jest uszkodzeniem jednego z czterech genów: BRAF znajdującego się na chromosomie 7q34, MAP2K1 znajdującego się na chromosomie 15q22, MAP2K2 znajdującego się na chromosomie 19p13 oraz KRAS znajdującego się na chromosomie 12p12[3]. Uszkodzenie genu BRAF występuje w 75% przypadkach zespołu sercowo-twarzowo-skórnego[3]. Wszystkie mutacje wpływają na szlak Ras/ERK (szlakMAPK/ERK), który ma znaczenie w regulacji wzrostu, podziału oraz śmierci komórek[4].
Epidemiologia
Częstość występowania szacowana jest na 1 na 810 000 żywych urodzeń[3]. Do 2015 opisano około 300 przypadków[3].
Obraz kliniczny
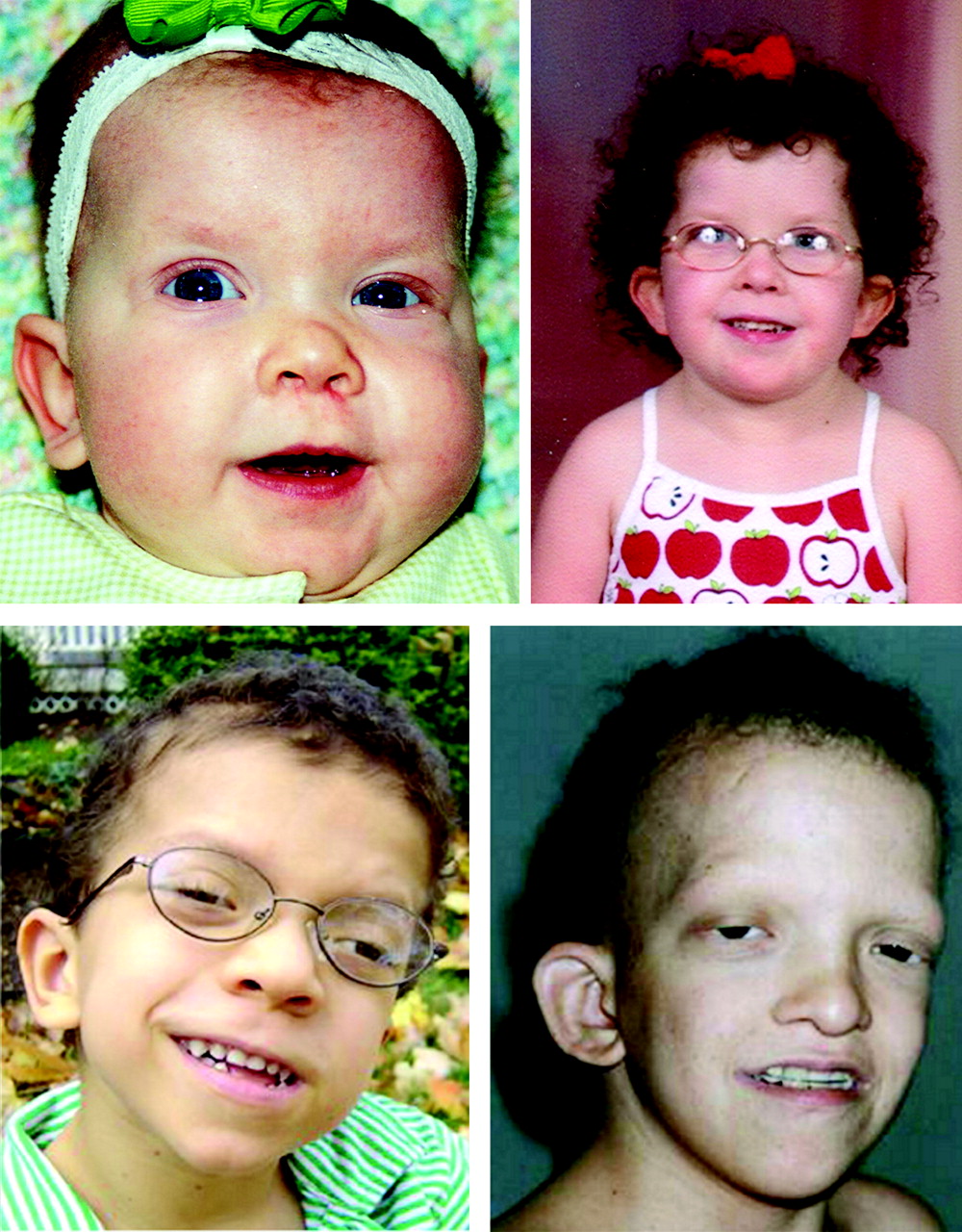
Większość pacjentów ma niedobór wzrostu ze względną makrocefalią[4]. Charakterystyczny jest typowy zespół zmian w zakresie twarzy i głowy: relatywnie duża głowa, szerokie czoło zwężające się ku skroniom, ptoza, krótki nos ze względnie szeroką podstawą, głęboka rynienka podnosowa z łukiem Kupidyna oraz małym podbródkiem[4]. Typowymi zmianami skóry i jej przydatków są rogowacenie mieszkowe oraz rzadkie, wolno rosnące, kręcone włosy[4]. Inne obserwowane zmiany skórne to: rybia łuska, rogowacenie mieszkowe, rogowacenie mieszkowe zanikowe twarzy, rogowiec dłoni i stóp[5]. Wada serca występuje u większości pacjentów[4]. Stwierdzane są ubytek w przegrodzie międzyprzedsionkowej, zwężenie drogi odpływu prawej komory oraz kardiomiopatia przerostowa[4].
Klasyfikacja
| Typ zespołu | uszkodzony gen |
|---|---|
| CFC typ I | BRAF |
| CFC typ II | KRAS |
| CFC typ III | MAP2K1 |
| CFC typ IV | MAP2K2 |
Typowe zmiany skórne występują u pacjentów z typem I zespołu, natomiast nie są obserwowane w typie II zespołu[6].
Diagnostyka różnicowa
Podstawą rozpoznania jest typowy obraz kliniczny oraz stwierdzenie mutacji w odpowiednim genie[7].
Leczenie
Nie ma leczenia przyczynowego zespołu sercowo-twarzowo-skórnego[4].
Rokowanie
Rokowanie u większości chorych co do życia jest dobre, jednakże ze względu na występujące wady serca długość życia może być skrócona[4].
Przypisy
- ↑
 N. Çelik, P. Cinaz, A. Bideci, Ö. Yüce i inni. Cardio-facio-cutaneous syndrome with precocious puberty, growth hormone deficiency and hyperprolactinemia.. „J Clin Res Pediatr Endocrinol”. 6 (1), s. 55-8, 2014. DOI: 10.4274/Jcrpe.1151. PMID: 24637312.
N. Çelik, P. Cinaz, A. Bideci, Ö. Yüce i inni. Cardio-facio-cutaneous syndrome with precocious puberty, growth hormone deficiency and hyperprolactinemia.. „J Clin Res Pediatr Endocrinol”. 6 (1), s. 55-8, 2014. DOI: 10.4274/Jcrpe.1151. PMID: 24637312. - ↑ JF. Reynolds, G. Neri, JP. Herrmann, B. Blumberg i inni. New multiple congenital anomalies/mental retardation syndrome with cardio-facio-cutaneous involvement--the CFC syndrome.. „Am J Med Genet”. 25 (3), s. 413-27, Nov 1986. DOI: 10.1002/ajmg.1320250303. PMID: 3789005.
- ↑ a b c d Cardiofaciocutaneous syndrome ORPHA1340 (ang.). Orphanet, 2015. [dostęp 2016-05-20].
- ↑ a b c d e f g h A. Roberts, J. Allanson, SK. Jadico, MI. Kavamura i inni. The cardiofaciocutaneous syndrome.. „J Med Genet”. 43 (11), s. 833-42, Nov 2006. DOI: 10.1136/jmg.2006.042796. PMID: 16825433.
- ↑ L. Borradori, C. Blanchet-Bardon. Skin manifestations of cardio-facio-cutaneous syndrome.. „J Am Acad Dermatol”. 28 (5 Pt 2), s. 815-9, May 1993. PMID: 8491871.
- ↑ a b Cardiofaciocutaneous syndrome 1; CFC1. Online Mendelian Inheritance in Man. [dostęp 2016-05-20].
- ↑ Agata Skórka, L. Korniszewski, D. Gieruszczak-Białek, J. Waligóra: W37 Zespół Nooan, zespół sercowo-twarzowo-skórny i zespół Costello – trudności diagnostyczne u dziecka z ciężką postacią zespołu Costello.. W: Polski Kongres Genetyki XV Zjazd Polskiego Towarzystwa Genetycznego IV Zjazd Polskiego Towarzystwa Genetyki Człowieka Warsztaty: Choroby genetyczne Gdańsk, 6-9 września 2004 r. Streszczenia. Gdańsk: 2004, s. 62. ISBN 83-920827-2-0. [dostęp 2016-05-20].
- ↑ a b Tomasz Floriańczyk, Agnieszka Tomik, Sylwia Łuszczyk, Małgorzata Gołąbek-Dylewska, Bożena Werner. Ocena przebiegu klinicznego wad serca u dzieci z zespołem Noonan. „Nowa Pediatria”, s. 122-127, 2014.
Linki zewnętrzne
- Ian Brown: The Boy in the Moon. [Historia rodziny chłopca z zespołem sercowo-twarzowo-skórnym ilustrowana 3 filmami] (ang.). The Globe an Mail. [dostęp 2016-05-20]. [zarchiwizowane z tego adresu (2016-05-02)].
- Zdjęcia twarzy dzieci z zespołem sercowo-twarzowo-skórnym (uwagę zwracają szerokie czoło, bulwiasty czubek nosa, nisko osadzone uszy, rzadkie włosy, brak brwi oraz rogowacenie mieszkowe czerwone bliznowaciejące) zdjęcie z artykułu: JF. Reynolds, G. Neri, JP. Herrmann, B. Blumberg i inni. New multiple congenital anomalies/mental retardation syndrome with cardio-facio-cutaneous involvement--the CFC syndrome.. „Am J Med Genet”. 25 (3), s. 413-27, Nov 1986. DOI: 10.1002/ajmg.1320250303. PMID: 3789005.
{kind=link}
![]() Przeczytaj ostrzeżenie dotyczące informacji medycznych i pokrewnych zamieszczonych w Wikipedii.
Przeczytaj ostrzeżenie dotyczące informacji medycznych i pokrewnych zamieszczonych w Wikipedii.
Media użyte na tej stronie

The Star of Life, medical symbol used on some ambulances.
Star of Life was designed/created by a National Highway Traffic Safety Administration (US Gov) employee and is thus in the public domain.Autor: Nurullah Çelik, Peyami Cinaz, Aysun Bideci, Özge Yüce, Hamdi Cihan Emeksiz, Esra Döğer, Orhun Çamurdan, Licencja: CC BY 2.5
Coarse facial appearance, curly and dry hair, sparse eyebrows, broad nasal tip with anteverted nares.